 Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)
Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)Qual a ação da substância do Tafinlar?
Resultados de Eficácia
Estudos clínicos
A eficácia deste medicamento no tratamento de pacientes adultos com melanoma irressecável ou metastático positivo para mutação BRAF V600 tem sido avaliada em 3 estudos (BRF113683 [BREAK-3], BRF113929 [BREAK-MB], e BRF113710 [BREAK-2]) incluindo pacientes com BRAF V600E e/ou mutações do V600K.
Pacientes não tratados previamente
A segurança e eficácia deste medicamento foram avaliadas em um estudo fase III, randomizado, aberto, [BREAK-3] comparando este medicamento a dacarbazina (DTIC) em pacientes com melanoma positivo para mutação BRAF V600E avançado (irressecável Estágio III) ou metastático (Estágio IV) não tratados previamente. A triagem incluiu teste central da mutação de BRAF V600E usando um ensaio de mutação de BRAF realizado na amostra do tumor mais recente disponível.
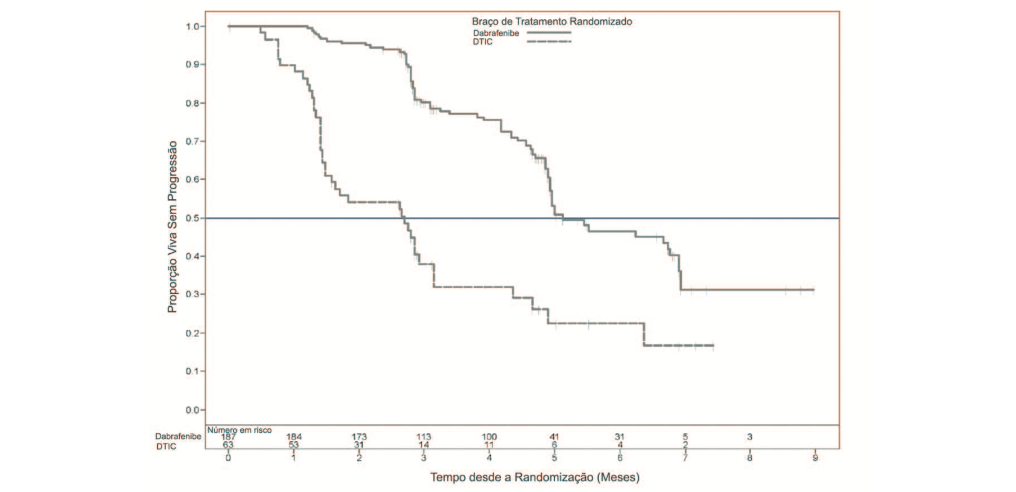
O estudo incluiu 250 pacientes randomizados 3:1 para receber ou este medicamento 150 mg duas vezes ao dia ou DTIC intravenoso 1000 mg/m2 a cada 3 semanas. O objetivo primário para este estudo era avaliar a eficácia deste medicamento comparado ao DTIC com relação à sobrevida livre de progressão (PFS) para pacientes com melanoma metastático com mutação positiva para BRAF V600E. Aos pacientes no braço do DTIC foi permitido receber este medicamento independente após confirmação radiográfica de progressão inicial. As características do período basal foram balanceadas entre os grupos de tratamento. Sessenta por cento dos pacientes eram homens e 99,6% eram caucasianos, a idade média era de 52 anos com 21% dos pacientes sendo ! 65 anos, 98,4% tinham condição de ECOG (Eastern Cooperative Oncology Group) de 0 ou 1, e 97% dos pacientes tinham doença metastática.
A análise primária foi baseada em 118 eventos no momento de corte de dados. A avaliação do investigador para os dados de eficácia estão resumidos na Tabela 1 e Figura 1.
Tabela 1: Dados de eficácia por avaliação do investigador para o Estudo BREAK-3
População com Intenção de Tratar | ||
Endpoints / Avaliação | este medicamento N=187 | DTIC N=63 |
Sobrevida livre de progressão (avaliação do investigador) | ||
Mediana, meses (95 % IC) HR (95 % IC) | 5,1 (4,9, 6,9) 2,7 (1,5, 3,2) 0,30 (0,18, 0,51) | |
Sobrevida Globala | ||
% até 6 meses (95 % IC) HR (95 % IC) | 87 (79,2, 91,9) 79 (59,7, 89,5) 0,61 (0,25, 1,48) | |
Resposta Globalb | ||
% (95 % IC)c | 53 (45,5, 60,3) | 19 (10,2, 30,9) |
Duração da resposta | ||
Mediana, meses (95 % IC) | N=99 | N=12 |
| Abreviações: IC: intervalo de confiança; DTIC: dacarbazina; HR: Hazard ratio; NR:não alcançado a Estimado a partir de curvas de Kaplan-Meier de 6 meses; Com a mediana de tempo de seguimento de 4,9 meses (alcance = 0 a 9,9 meses) e 30 mortes, dados de sobrevida global ainda não estão maduros e mediana de sobrevida global não foi atingida por nenhum dos braços. Indivíduos são resumidos pelo tratamento randomizado; as estimativas incluem dados da fase de cruzamento para indivíduos randomizados para DTIC e, portanto, reflete qualquer benefício de segunda linha deste medicamento. b Definida como resposta completa+resposta parcial. c Resposta confirmada. | ||
Vinte e oito indivíduos (44 %) randomizados para DTIC cruzaram para este medicamento seguindo a progressão da doença verificada de forma independente. O tempo mediano neste medicamento após o cruzamento foi de 2,8 meses e a taxa de resposta global (ORR) não confirmada foi de 46%.
Figura 01: Avaliação Kaplan-Meier do investigador da sobrevida livre de progressão– pacientes não tratados previamente (população ITT)
Pacientes com metástases cerebrais
BREAK-MB foi um estudo multicêntrico, aberto, de duas coortes, de Fase II desenhado para avaliar a resposta intracranial deste medicamento em indivíduos com confirmação histológica (Estágio IV) de melanoma com mutação positiva BRAF (V600E ou V600K) metastático para o cérebro. Os indivíduos foram incluídos na Coorte A (indivíduos sem tratamento local prévio para metástases cerebrais) ou Coorte B (indivíduos que receberam tratamento local prévio para metástases cerebrais). Os resultados estão resumidos na Tabela 02.
Tabela 2: Dados de eficácia em pacientes com metástases no cérebro (Estudo BREAK-MB)
Toda População de Indivíduos Tratados | ||||
BRAF V600E (Primário) | BRAF V600K | |||
Endpoints / Avaliação | Coorte A N=74 | Coorte B N=65 | Coorte A N=15 | Coorte B N=18 |
Taxa de resposta intracraniana global, % (95 % IC)a | ||||
39% (28,0, 51,2) P < 0,001b | 31% (19,9, 43,4) P < 0,001b | 7% (0,2, 31,9) | 22% (6,4, 47,6) | |
Duração da resposta intracraniana, mediana, meses (95% IC) | ||||
N=29 | N=20 | N=1 | N=4 | |
Resposta Global, % (95% IC)a | ||||
38% (26,8, 49,9) | 31% (19,9, 43,4) | 0 (0, 21,8) | 28% (9,7, 53,5) | |
Duração da resposta, mediana, meses (95% IC) | ||||
N=28 | N=20 | NA | N=5 | |
Sobrevida livre de progressão, mediana, meses (95% IC) | ||||
3,7 (3,6, 5,0) | 3,8 (3,6, 5,5) | 1,9 (0,7, 3,7) | 3,6 (1,8, 5,2) | |
Sobrevida Global, mediana, meses (95% IC) | ||||
Mediana, meses | 7,6 (5,9, NR) | 7,2 (5,9, NR) | 3,7 (1,6, 5,2) | 5,0 (3,5, NR) |
| Abreviações: IC: intervalo de confiança; NR: não alcançado; NA: não aplicável a – Resposta confirmada. b – Este estudo foi desenhado para apoiar ou rejeitar a hipótese nula de OIRR% ≤ 10 (com base nos resultados históricos) em favor da hipótese alternativa de OIRR ≥ 30% em indivíduos BRAF V600E positivos. | ||||
Pacientes que não foram tratados previamente ou falharam em pelo menos uma terapia sistêmica prévia
BRF113710 (BREAK-2) foi um estudo multicêntrico, global, aberto, braço único, de Fase II que incluiu 92 indivíduos de pesquisa com melanoma metastático confirmado histologicamente (Estágio IV) com melanoma positivo para mutação BRAF V 600E ou V600K confirmada. Os indivíduos eram virgens de tratamento (n=15) ou receberam tratamento prévio (n=77) na presença de metástases (por exemplo, quimioterapia, imunoterapia, terapia alvo prévia, etc.).
O investigador avaliou a taxa de resposta confirmada na eficácia primária em uma população de pacientes com melanoma metastático BRAF V600E (n=76) foi de 59 % (95% IC: 48,2; 70,3) incluindo 7% de resposta completa. A mediana de PFS foi de 6,3 meses (95% IC: 4,6; 7,7) e a duração mediana da resposta foi de 5,2 meses (95 % IC: 3,9, não calculado). A terapia sistêmica prévia não pareceu afetar significativamente a resposta. O investigador avaliou a taxa de resposta confirmada na eficácia secundária em uma população de pacientes com melanoma metastático com mutação positiva BRAF V600K (n=16) foi de 13% (95% IC: 0,0; 28,7) com duração mediana da resposta de 5,3 meses (95 % IC: 3,7; 6,8). Não houve resposta completa na população de pacientes V600K.
Características Farmacológicas
Propriedades Farmacodinâmicas
Mecanismo de ação
O dabrafenibe é um inibidor de RAF quinase ATP-competitivo, potente e seletivo, com valores de IC50 de 0,65, 0,5 e 1,84 nM para as enzimas BRAF V600E, BRAF V600K e BRAF V600D, respectivamente. As mutações oncogênicas em BRAF levam a ativação constitutiva da via RAS/RAF/MEK/ERK e estimulação do crescimento das células tumorais. As mutações de BRAF têm sido identificadas em uma alta frequência em cânceres específicos, incluindo aproximadamente 50% dos melanomas. A mutação de BRAF mais comumente observada, V600E, e a próxima mais comum, V600K, respondem por 95% das mutações de BRAF encontradas em todos os pacientes com câncer. Um número de substituições raras também ocorre incluindo V600D, V600G e V600R.
O dabrafenibe também inibe BRAF selvagem e enzimas CRAF com valores de IC50 DE 3,2 E 5,0 nM, respectivamente. O dabrafenibe inibe o crescimento celular de melanoma mutante BRAF V600 in vitro e in vivo.
Efeitos Farmacodinâmicos
O dabrafenibe demonstrou supressão de um biomarcador farmacodinâmico à jusante (ERK fosforilado) em linhagens celulares de melanoma mutante BRAF V600, in vitro e em modelos animais.
Em indivíduos com melanoma mutante BRAF V600, a administração deste medicamento resultou em inibição de ERK fosforilado do tumor em relação ao período basal.
Eletrofisiologia cardíaca: O efeito potencial do dabrafenibe no prolongamento QT foi avaliado em um estudo QT com múltiplas doses. Uma dose supraterapêutica de 300 mg de dabrafenibe duas vezes ao dia foi administrada em 32 indivíduos com tumores com mutação BRAF- V600 positiva. Nenhum evento clinicamente relevante de dabrafenibe ou seus metabólitos no intervalo QTc foi observado.
Propriedades Farmacocinéticas
Absorção
O dabrafenibe é absorvido oralmente com tempo mediano para alcançar o pico de concentração plasmática de 2 horas após a dose. A biodisponibilidade absoluta média de dabrafenibe oral é de 95 % (90% IC: 81;110). A exposição ao dabrafenibe (Cmáx e AUC) aumentou de uma forma proporcional à dose entre 12 e 300 mg seguindo a administração de dose única, mas o aumento foi menor que proporcional a dose após repetir a dose duas vezes ao dia. Houve uma diminuição na exposição observada com a repetição da dose, provavelmente devido à indução de seu próprio metabolismo. A razão de acumulação média de AUC dia 18/Dia 1 foi 0,73. Após administração de 150 mg duas vezes ao dia, a média geométrica Cmáx, AUC(0-t) e a concentração pré-dose (Ct) foram 1478 ng/mL, 4341 ng*hr/mL e 26 ng/mL, respectivamente.
A administração deste medicamento com comida reduziu a biodisponibilidade (Cmáx e AUC diminuíram para 51% e 31% respectivamente) e retardou a absorção de dabrafenibe em cápsulas, quando comparado ao estado de jejum.
Distribuição
O dabrafenibe liga-se a proteína plasmática humana e é 99,7% ligado. A distribuição do volume em estado de equilíbrio após administração intravenosa de microdose é 46 L.
Biotransformação/Metabolismo
O metabolismo de dabrafenibe é mediado principalmente por CYP2C8 e CYP3A4 para formar hidroxi-dabrafenibe, o qual é posteriormente oxidado via CYP3A4 para formar carboxi-dabrafenibe. Carboxi-dabrafenibe pode ser descarboxilado via um processo não enzimático para formar desmetil-dabrafenibe. Carboxi-dabrafenibe é excretado na bile e urina. Desmetil-dabrafenibe também pode ser formado no intestino e reabsorvido. Desmetil-dabrafenibe é metabolizado pelo CYP3A4 a metabólitos oxidativos. A meia-vida terminal de hidroxi-dabrafenibe corresponde àquela do parental com meia-vida de 10 horas enquanto que os metabólitos carboxi- e desmetil- exibiram meia-vidas mais longas (21-22 horas). As médias da razão de AUC parental do metabólito após a administração de dose repetida foram 0,9, 11 e 0,7 para hidroxi-, carboxi- e desmetil-dabrafenibe, respectivamente. Com base na exposição, potência relativa e propriedades farmacocinéticas, tanto para hidroxi- e desmetil-dabrafenibe são passíveis de contribuir para a atividade clínica deste medicamento, enquanto que a atividade do carboxi- dabrafenibe não é passível de ser significativa.
Eliminação
A meia-vida terminal seguida de microdose IV é de 2,6 horas. A meia-vida terminal de dabrafenibe é de 8 horas devido a uma fase terminal prolongada após a administração oral. O clearance plasmático IV é 12 L/hr.
A excreção fecal é a rota principal de eliminação após a administração oral, representando 71 % da dose radioativa enquanto a excreção urinária representa 23 % da radioatividade.
Populações Especiais
Insuficiência Hepática
A farmacocinética de dabrafenibe foi caracterizada em 65 pacientes com insuficiência hepática leve (com base na classificação do Instituto Nacional do Câncer [NCI]) incluídos em estudos clínicos, utilizando uma análise da população. O clearance do dabrafenibe oral não foi significativamente diferente entre estes indivíduos e indivíduos com função hepática normal (4% de diferença). Além disso, insuficiência hepática leve não teve um efeito significante na concentração plasmática do metabólito de dabrafenibe. Não há dados disponíveis em pacientes com insuficiência hepática moderada a grave (ver Posologia e Modo de Usar).
Insuficiência Renal
A farmacocinética de dabrafenibe foi caracterizada em 233 pacientes com insuficiência renal leve (GFR 60-89 mL/min/1,73 m2) e em 30 pacientes com comprometimento renal moderado (GFR 30-59 mL/min/1,73 m2) incluídos em estudos clínicos utilizando análise de população. O efeito da insuficiência renal leve ou moderada no clearance oral de dabrafenibe foi pequeno < 6%, para ambas as categorias) e não foi clinicamente relevante. Além disso, insuficiência renal leve ou moderada não teve efeito significativo nas concentrações plasmáticas de hidroxi-, carboxi- e desmetil-dabrafenibe. Não existem dados disponíveis em indivíduos com insuficiência renal grave (ver Posologia e Modo de Usar).
Idade
Baseado na análise farmacocinética de população, a idade não tem nenhum efeito significativo na farmacocinética de dabrafenibe. Idade superior a 75 anos foi um preditor significativo das concentrações plasmáticas de carboxi- e desmetil-dabrafenibe com uma exposição 40% maior em indivíduos ≥ 75 anos de idade, em relação aos indivíduos <75 anos de idade.
Peso corporal e Sexo
Baseados na análise farmacocinética de população, sexo e peso corporal mostraram influenciar o clearance oral de dabrafenibe; o peso também impactou a distribuição do volume oral e o clearance distributivo. Estas diferenças farmacocinéticas não foram consideradas clinicamente relevantes.
Raça/Etnia
A análise farmacocinética da população não mostrou diferenças significativas na farmacocinética de dabrafenibe entre pacientes asiáticos e caucasianos. Nenhum ajuste de dose é necessário em pacientes asiáticos.
Não há dados suficientes para avaliar o potencial efeito da raça sobre a farmacocinética de dabrafenibe.
Avaliação in vitro do potencial de interações medicamentosas
Efeito deste medicamento em outros fármacos
Em hepatócitos humanos, dabrafenibe produziu aumento concentração-dependente nos níveis de RNAm de CYP2B6 e CYP3A4 em até 32 vezes os níveis de controle.
Embora dabrafenibe e seus metabólitos hidroxi-dabrafenibe, carboxi-dabrafenibe e desmetil-dabrafenibe, sejam inibidores do polipeptídio transportador de ânions orgânicos humanos (OATP) 1B1, OATP1B3, transportador de ânion orgânico (OAT)1 e AOT3 e devido ao fato que dabrafenibe e seu metabólito desmetil demonstraram serem inibidores do transportador de cátions orgânicos 2 (OCT2) in vitro, o risco de interação medicamentosa é mínimo com base na exposição clínica.
O dabrafenibe e desmetil-dabrafenibe demonstraram serem inibidores moderados da BCRP; porém, com base na exposição clínica, o risco de interação medicamentosa é mínimo.
Nem dabrafenibe ou seus 3 metabólitos demonstraram ser inibidores da Pgp in vitro.
Efeito de outros fármacos neste medicamento:
Resultados de estudos in vitro indicam que CYP2C8 e CYP3A4 são as principais enzimas CYP envolvidas no metabolismo oxidativo do dabrafenibe, enquanto hidroxi-dabrafenibe e desmetil-dabrafenibe são substratos do CYP3A4. Portanto, inibidores ou indutores dessas enzimas têm o potencial de afetar a farmacocinética do dabrafenibe ou de seus metabólitos (ver Interações Medicamentosas).
O dabrafenibe é um substrato da Pgp humana e da Proteína de Resistência ao Câncer de Mama (BCRP)1 in vitro. Entretanto, estes transportadores tiveram um impacto mínimo na biodisponibilidade oral de dabrafenibe e na eliminação, e o risco de interação medicamentosa é mínimo.