 Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)
Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)Qual a ação da substância do Starform?
Resultados da eficácia
Um estudo de 24 semanas duplo-cego, randomizado, grupo paralelo, de dose fixa foi feito para avaliar a segurança, eficácia e tolerabilidade da nateglinida em combinação com metformina comparada com a metformina em monoterapia em indivíduos com diabetes mellitus não insulino-dependentes inadequadamente controlada com meformina e dieta.
Pacientes do sexo masculino e feminino com 30 anos ou mais com diabetes mellitus Tipo 2 há pelo menos 6 meses, que receberam metformina em monoterapia por um período mínimo de 3 meses, nos quais a dose diária de metformina foi ≥ 1500 mg por no mínimo um mês antes da semana -4, foram incluídos. O estudo consistiu em um período de entrada (“run-in”) simples-cego de 4 semanas e de um período de tratamento de 24 semanas duplo-cego randomizado. Durante o período run-in, todos os pacientes receberam nateglinida placebo antes das três refeições principais e 1.000 mg de metformina duas vezes por dia (antes ou imediatamente após o café da manhã e jantar).
Todos os pacientes randomizados continuaram a receber metformina 1.000 mg duas vezes (antes ou imediatamente após o café da manhã e jantar) e, adicionalmente foram randomizados para receber nateglinida placebo (152 pacientes), 60 mg (155 pacientes) ou 120 mg (160 pacientes) antes das três refeições principais. O critério chave de inclusão para o período duplo-cego, randomizado foi uma HbA1C média das visitas das semanas -4 e -2 na faixa de 6,8-11,0%. Pacientes com glicemia plasmática de jejum (GPJ) > 15 mmol/L na semana -4 ou na semana -2 foram excluídos.
Os pacientes foram avaliados pela variação da HbA1C, glicemia de jejum, peso corporal e perfil lipídico iniciais. As variáveis de segurança foram os eventos adversos, a automonitorização da glicose sanguínea (AMGS) para hipoglicemia suspeita, sinais vitais, avaliações de ECG e avaliações laboratoriais (hematologia, química e exame de urina).
Reduções estatisticamente significativas na HbA1C e GPJ iniciais foram observadas em pacientes tratados com terapia combinada em comparação com metformina em monoterapia (veja Tabelas 1 e 2). Não foram observadas alterações significativas em nenhum dos grupos de tratamento para a mudança no peso corporal e perfil lipídico.
Tabela 1 - HbA1C (%) inicial e alterações ajustadas desde o início até o desfecho do estudo populacional (IDT)
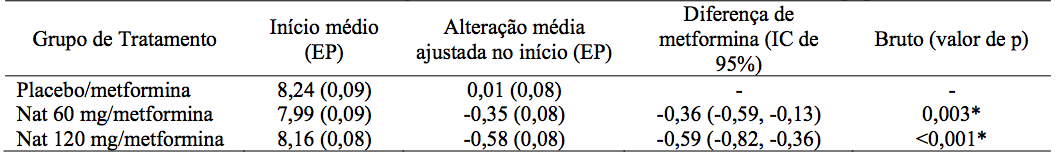
*Estatisticamente significante no nível de 0,05 após o ajuste de Dunnett para comparações múltiplas.
Ajustados = mínimos quadrados médios.
EP = erro padrão.
IC = intervalo de confiança.
Nat = nateglinida.
Tabela 2 - GPJ (mmol/l) inicial e alterações ajustadas desde o início até o desfecho do estudo populacional (IDT)
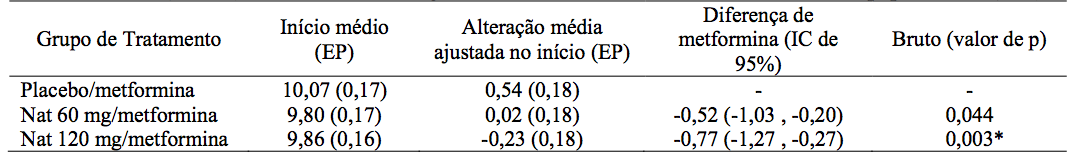
*Estatisticamente significante no nível de 0,05 após o ajuste de Dunnett para comparações múltiplas Ajustados = mínimos quadrados médios.
EP = erro padrão.
IC = intervalo de confiança.
Nat = nateglinida.
O estudo de 24 semanas, duplo-cego randomizado, grupo paralelo foi conduzido para avaliar prospectivamente a eficácia, segurança e tolerabilidade da nateglinida em monoterapia, metformina em monoterapia, nateglinida em combinação com metformina e placebo em indivíduos com diabetes mellitus não insulino-dependente inadequadamente controlada apenas com dieta. Paciente do sexo masculino e feminino com 30 anos ou mais com diabetes mellitus Tipo 2 há pelo menos 3 meses, que estava em dieta por 3 semanas antes da visita da semana -4 foram avaliados para a mudança no HbA1c inicial.
Foi prescrito um comprimido de 120 mg de nateglinida antes do café da manhã, almoço e jantar. Foi prescrito metformina com ou imediatamente após o café da manhã (semana 1), com ou imediatamente após o café da manhã e jantar (semana 2) e com ou imediatamente após as três refeições (semana 3 em diante).
Alteração na glicemia plasmática de jejum (GPJ), peso corporal, qualidade de vida (QDV), perfil lipídico, glicemia pré e pós-Sustacal, insulina e C-peptídeo. Parâmetro primário de eficácia foi avaliado usando a intenção de tratar (IDT). O modelo ANCOVA, ajustando para o tratamento, inicial, centro, tratamento por centro e tratamento por interação inicial foi usado para analisar os desfechos primário e secundário.
505 pacientes completaram o estudo (135 com nateglinida + metformina, 133 com nateglinida, 132 com metformina e 105 com placebo). Alteração no HbA1C produziu no desfecho de nateglinida (-0,45%) e metformina (-0,78%) parece ser sinérgica, produzindo um decréscimo de 1,43% para a terapia combinada (veja Tabela 3).
Tabela 3 - Alterações no início em HbA1C (%) até o desfecho do estudo populacional (IDT)
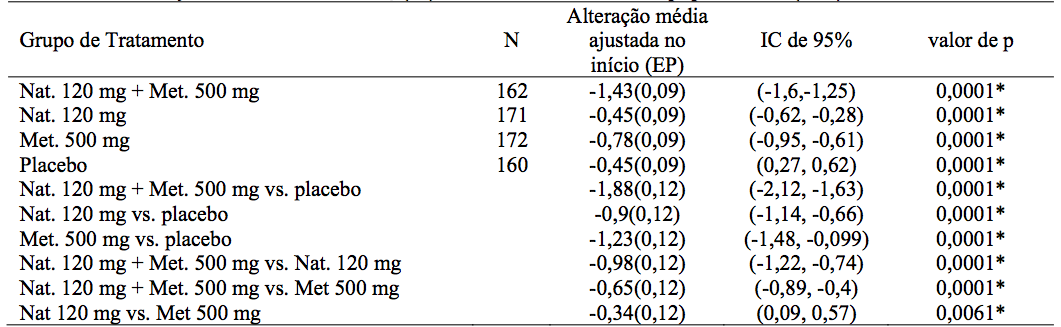
* Indica significância estatística no nível de 0,05.
Nat = nateglinida, Met = metformina.
EP = erro padrão.
IC = intervalo de confiança.
A redução no GPJ para a terapia de nateglinida em combinação com metformina foi estatisticamente significante maior do que nateglinida e metformina em monoterapia, e a redução para a metformina em monoterapia foi significante maior do que nateglinida em monoterapia (veja Tabela 4).
Tabela 4 - Alterações de GPJ (mmol/L) do início ao desfecho do estudo (IDT populacional)
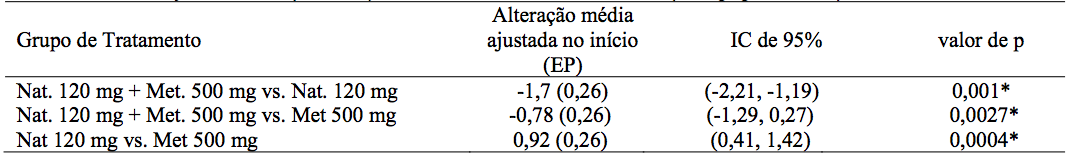
* Indica significância estatística no nível de 0,05.
Nat = nateglinida, Met = metformina.
EP = erro padrão.
IC = intervalo de confiança.
A terapia de combinação e a nateglinida em monoterapia foram significantemente melhores do que o placebo e a metformina em monoterapia na diminuição das excursões de glicose pós-Sustacal pós-prandial.
Após 24 semanas de tratamento, pacientes que completaram o estudo principal ou descontinuaram por sintomas de hiperglicemia ou resposta terapêutica insatisfatória após pelo menos 12 semanas de tratamento foram inscritos em um estudo de extensão duplo-cego, randomizado, grupo paralelo, dose fixa de 28 semanas de duração. Deste modo, 52 semanas do total do vínculo de tratamento duplo foi administrado desde a randomização no estudo principal. Os indivíduos elegíveis continuaram o tratamento duplo-cego randomizado em toda a extensão, com uma exceção: indivíduos no grupo de tratamento duplo-cego com placebo foram transferidos para o grupo duplo-cego com nateglinida em consequência da entrada neste estudo de extensão. 400 indivíduos (103 no grupo com nateglinida mais metformina, 104 no grupo com nateglinida, 89 no grupo com nateglinida placebo e 104 no grupo com metformina) foram inscritos no estudo de extensão e 224 completaram o estudo (69 no grupo com nateglinida mais metformina, 48 no grupo com nateglinida, 47 no grupo com nateglinida placebo e 60 no grupo com metformina).
A terapia de combinação de nateglinida e metformina resultou em uma redução significantemente maior na HbA1C do início para a extensão da intenção de tratar (EXT IDT) da população em relação a qualquer uma das monoterapias e placebo. No entanto, a redução foi significativa em GPJ com terapia combinada apenas quando em comparação com nateglinida monoterapia e com placebo.
A nateglinida e metformina parecem ter mecanismo de ação complementar quando administrados com uma terapia de combinação, com efeitos da metformina sobre o GPJ e efeitos da nateglinida sobre as excursões de glicose pós-prandial produzindo diminuição sinérgica na HbA1C e GPJ.
Características Farmacológicas
Grupo farmacoterapêutico: outros medicamentos hipoglicemiantes orais.
Código ATC: Nateglinida: A10BX03 e Metformina: A10BA02.
Mecanismo de ação/Farmacodinâmica
Nateglinida
A nateglinida é um derivado de um aminoácido (fenilalanina), que é química e farmacologicamente diferente de outros agentes antidiabéticos. Restaura a secreção primária de insulina, resultando em uma redução da glicose pós-prandial e HbA1C.
A secreção primária de insulina é um mecanismo essencial para a manutenção do controle glicêmico normal. A nateglinida, quando tomada antes das refeições, restaura a fase primária ou primeira fase de secreção de insulina, que é perdida em pacientes com diabetes Tipo 2. Esta ação é mediada por uma interação rápida e transitória com canal de íon potássio-ATP (K+ATP) nas células beta do pâncreas. Estudos eletrofisiológicos demonstraram que a nateglinida tem uma seletividade 300 vezes maior para as células beta do pâncreas em relação aos canais de K+ATP cardiovasculares.
Ao contrário de outros agentes antidiabéticos orais, a nateglinida induz uma significativa secreção de insulina durante os primeiros 15 minutos após uma refeição. Isto reduz as excursões de glicemia pós-prandial (picos). Os níveis de insulina retornam aos valores basais em 3 a 4 horas, reduzindo a hiperinsulinemia pós-prandial, a qual tem sido associada com hipoglicemia tardia. A nateglinida é rapidamente eliminada.
A secreção induzida pela nateglinida de insulina pelas células beta do pâncreas é sensível à glicose, de tal forma que é secretada menos insulina à medida que o nível de glicose cai. Inversamente, a coadministração de alimentos ou infusão de glicose resulta em um claro aprimoramento da secreção de insulina. O reduzido potencial da nateglinida em estimular a secreção de insulina em baixas concentrações de glicose no ambiente proporciona proteção adicional à hipoglicemia, tal como quando se deixa de ingerir uma refeição.
Cloridrato de metformina
O cloridrato de metformina é um antidiabético oral pertencente ao grupo químico das biguanidas.
Em contraste com as sulfonilureias, o cloridrato de metformina não estimula a secreção da insulina e não tem efeito hipoglicemiante em não diabéticos. Em diabéticos, o cloridrato de metformina diminui a hiperglicemia com baixo risco de causar episódios hipoglicêmicos (exceto em jejum prolongado ou em combinação com sulfonilureias ou insulina).
O mecanismo de ação do cloridrato de metformina é caracterizado por:
- Um aumento da sensibilidade periférica à insulina e da absorção celular de glicose;
- Uma inibição da gliconeogênese hepática;
- Um retardo da absorção intestinal de glicose.
Estas ações combinadas contribuem para que o cloridrato de metformina reduza a hiperglicemia e melhore a tolerância à glicose. A ação periférica do cloridrato de metformina sobre a resistência à insulina é provavelmente acompanhada por um efeito pós-receptor, independente da ligação receptor-insulina.
Em estudos com cloridrato de metformina um efeito favorável foi observado sobre o metabolismo de lipídeos (especialmente uma redução dos níveis aumentados de colesterol total e em alguns estudos também uma redução dos níveis aumentados de triglicérides).
Farmacocinética
Nateglinida
Absorção
A nateglinida é rapidamente absorvida após a administração oral do comprimido antes de uma refeição, com o pico de concentração média da nateglinida ocorrendo geralmente em menos de uma hora. A nateglinida é rápida e quase completamente absorvida ( maior ou igual a 90%) a partir de uma solução oral. Calcula-se que a biodisponibilidade oral absoluta seja de 72%. Em pacientes diabéticos Tipo 2, aos quais foi administrada nateglinida no intervalo de doses de 60 a 240 mg antes das três refeições por dia, durante uma semana, a nateglinida apresentou uma farmacocinética linear tanto para a área sob a curva (ASC) quanto para a Cmáx, e o Tmáx foi independente da dose.
Distribuição
Calcula-se que o volume de distribuição da nateglinida em estado de equilíbrio, com base em dados intravenosos, seja de aproximadamente 10 litros. Estudos in vitro mostraramque a nateglinida está extensivamente ligada (97-99%) às proteínas plasmáticas, principalmente à albumina sérica e, em menor extensão, à alfa1-ácido-glicoproteína. A extensão da ligação às proteínas séricas é independente da concentração do fármaco no intervalo de teste de 0,1-10 micrograma de nateglinida/mL.
Biotransformação/Metabolismo
A nateglinida é extensivamente metabolizada pelo sistema oxidase de função mista antes da eliminação. Os principais metabólitos encontrados em humanos resultam da hidroxilação da cadeia lateral isopropil, ou do carbono metil ou em um dos grupos metil. A atividade dos principais metabólitos écerca de 5-6 e 3 vezes menos potente do que a da nateglinida, respectivamente. Os metabólitos menores identificados foram um diol, um isopropeno e acilglicuronídio(s) da nateglinida. Apenas o metabólito menor isopropeno possui atividade, que é quase tão potente quanto a nateglinida.
Dados disponíveis de experimentos in vitro e in vivo indicam que a nateglinida é predominantemente metabolizada pelo citocromo P450 2C9 (70%) e em menor grau pelo CYP 3A4 (30%).
Eliminação
A nateglinida e os seus metabólitos são rápida e completamente eliminados.
Aproximadamente 75% da nateglinida [14C] administrada é recuperada na urina em seis horas após a dose. A maioria da nateglinida [14C] é excretada através da urina (83%), com um adicional de 10% eliminado nas fezes. Aproximadamente 6-16% da dose foi excretada na urina como fármaco inalterado. As concentrações plasmáticas diminuem rapidamente e a meia-vida de eliminação da nateglinida foi, em geral, por volta de 1,5 hora em todos os estudos da nateglinida com voluntários e pacientes diabéticos Tipo 2.
De forma consistente com a sua curta meia-vida de eliminação, não há acúmulo aparente de nateglinida com doses múltiplas de até 240 mg três vezes diariamente.
Efeito dos alimentos
Quando da administração pós-prandial, a absorção da nateglinida (ASC) permanece inalterada. No entanto, há um atraso na taxa de absorção caracterizado por uma diminuição na Cmáx e um atraso no tempo para a concentração plasmática máxima (Tmáx). É recomendado que a nateglinida seja administrada antes das refeições. Normalmente é ingerida imediatamente (1 minuto) antes de uma refeição, mas pode ser ingerida até 30 minutos antes das refeições.
População especial
Gênero:
Não foram observadas diferenças clinicamente significativas na farmacocinética da nateglinida entre homens e mulheres.
Idade:
A idade não influenciou as propriedades farmacocinéticas da nateglinida.
Insuficiência hepática:
A disponibilidade sistêmica e a meia-vida da nateglinida em indivíduos não diabéticos com insuficiência hepática leve a moderada, não diferem de forma clinicamente significativa das dos indivíduos saudáveis. Os pacientes com doença hepática grave não foram estudados, e neste grupo, a nateglinida deve ser utilizada com precaução.
Insuficiência renal:
A disponibilidade sistêmica e a meia-vida da nateglinida em indivíduos diabéticos com insuficiência renal moderada a grave [clearance (depuração) de creatinina 15-50 mL/min/1,73 m2] e em pacientes que necessitam de diálise, não diferem de forma clinicamente significativa das dos indivíduos saudáveis.
Cloridrato de metformina
Absorção
Após uma dose oral de cloridrato de metformina, o pico de concentração plasmática (Cmax) é alcançado em 2,5 horas (Tmáx). A biodisponibilidade absoluta dos comprimidos de 500 ou 850 mg de cloridrato de metformina é de aproximadamente 50-60% em indivíduos saudáveis.
Após administração oral, a absorção de cloridrato de metformina é saturável e incompleta. Assume-se que a farmacocinética da absorção de cloridrato de metformina é não linear.
Nas doses e nos esquemas de dosagem habituais, as concentrações plasmáticas do cloridrato de metformina no estado de equilíbrio são alcançadas dentro de 24 a 48 horas e são geralmente menores do que 11 microgramas/mL. Em ensaios clínicos controlados, os níveis plasmáticos máximos de cloridrato de metformina (Cmáx) não excederam 4 microgramas/mL, mesmo com doses máximas.
A alimentação diminui a extensão e atrasa levemente a absorção de cloridrato de metformina, como demonstrado pela Cmax aproximadamente 40% menor, uma ASC (área sob a curva) 25% menor e um prolongamento de 35 minutos no Tmax após a administração de dose única de um comprimido de 850 mg do cloridrato de metformina com alimento, comparado com o comprimido na mesma concentração administrado em jejum. A relevância clínica destas reduções é desconhecida.
Distribuição
O volume de distribuição aparente do cloridrato de metformina após doses orais únicas de 850 mg foi em média 654 ± 358 L. O cloridrato de metformina liga-se de forma não significativa às proteínas plasmáticas, em contraste com as sulfonilureias, que são mais de 90% ligadas às proteínas. Partições do cloridrato de metformina nos eritrócitos, a maioria provavelmente em função do tempo. Em doses clínicas usuais e esquemas de dosagem do cloridrato de metformina, as concentrações plasmáticas no estado de equilíbrio do cloridrato de metformina são atingidas dentro de 24 a 48 horas e são geralmente menos de 1 micrograma/mL.
Biotransformação/Metabolismo
O cloridrato de metformina é excretado inalterado na urina. Nenhum metabólito foi identificado em humanos.
Eliminação
Após uma dose oral, a fração não absorvida recuperada na fezes foi 20-30%.
O clearance renal (depuração) do cloridrato de metformina é maior que 400 mL/min, 3,5 vezes maior que o clearance (depuração) da creatinina, o que indica que a secreção tubular é a principal via de eliminação.
Após a administração oral, aproximadamente 90% do fármaco absorvido é eliminado pela via renal dentro das primeiras 24 horas, com uma meia-vida de eliminação plasmática de aproximadamente 6,2 horas. No sangue, a meia-vida de eliminação é aproximadamente 17,6 horas, sugerindo que a massa dos eritrócitos possa ser um compartimento de distribuição.
População especial
Idade:
A idade (12-45 anos) não influenciou as propriedades farmacocinéticas da metformina. No entanto, o clearance (depuração) da metformina pode ser reduzido em pacientes idosos com função renal comprometida.
Gênero:
Parâmetros farmacocinéticos do cloridrato de metformina não diferem significativamente entre os indivíduos normais e pacientes com diabetes Tipo 2, quando analisados de acordo com o sexo (homens = 19, mulheres = 16). Da mesma forma, em estudos clínicos controlados em pacientes com diabetes do Tipo 2, o efeito anti-hiperglicemiante do cloridrato de metformina foi comparável em homens e mulheres.
Insuficiência hepática:
Não foram conduzidos estudos de farmacocinética do cloridrato de metformina em indivíduos com insuficiência hepática.
Insuficiência renal:
Em pacientes com insuficiência renal, o clearance (depuração) renal da metformina é diminuído proporcionalmente ao clearance (depuração) da creatinina e assim, a meia-vida de eliminação é prolongada, conduzindo ao aumento dos níveis plasmáticos de cloridrato de metformina.
Sensibilidade étnica:
Não foram realizados estudos dos parâmetros de corrida farmacocinética do cloridrato de metformina. No entanto, em estudos clínicos controlados do cloridrato de metformina em pacientes com diabetes do Tipo 2, o efeito anti-hiperglicêmico foi comparável em brancos (n = 249), negros (n = 51) e hispânicos (n = 24).
Dados de segurança pré-clínicos
Não foram realizados estudos pré-clínicos com a combinação de nateglinida e cloridrato de metformina. Assim, a informação sobre a segurança pré-clínica deste produto é baseada nos dados obtidos a partir dos componentes individuais.
Nateglinida
Os dados pré-clínicos não revelaram perigo especial para os humanos, com base nos estudos convencionais de toxicidade de doses repetidas.
Mutagenicidade
Uma avaliação da nateglinida em uma série de testes padrões de genotoxicidade in vitro e in vivo, não revelaram qualquer potencial genotóxico.
Carcinogenicidade
Os estudos de carcinogenicidade realizados em camundongos e ratos sugerem que a administração oral de nateglinida não está associada a qualquer lesão neoplásica.
Toxicidade reprodutiva e no desenvolvimento
Dados pré-clínicos indicaram que não há perigo especial na fertilidade e função reprodutiva.
Durante o período pós-parto, o peso corporal foi menor na prole de ratos administrados com nateglinida 1.000 mg/kg (aproximadamente 60 vezes a exposição terapêutica humana com uma dose recomendada de nateglinida de 120 mg, três vezes ao dia, antes das refeições). A nateglinida não foi teratogênica em ratos. Em coelhos, o desenvolvimento embrionário foi afetado negativamente e a incidência de agenesia da vesícula biliar ou pequena vesícula biliar, foi aumentada com doses de 300 e 500 mg/kg.
A dose de 500 mg/kg também foi associada com toxicidade materna em coelhos. Estudos em ratos não demonstraram efeito no parto com doses até 1.000 mg/kg (aproximadamente 60 vezes a exposição terapêutica humana com uma dose máxima recomendada de nateglinida 120 mg, três vezes ao dia, antes das refeições).
Cloridrato de metformina
Dados pré-clínicos não revelaram perigo especial para o ser humano, segundo estudos convencionais de toxicidade de dose repetida com cloridrato de metformina.
Mutagenicidade
Uma avaliação do cloridrato de metformina em uma série de testes padrões de genotoxicidade in vitro e in vivo, não revelaram qualquer potencial genotóxico.
Carcinogenicidade
Foram realizados estudos de carcinogenicidade de longa duração com o cloridrato de metformina em ratos (dosagem de duração 104 semanas) e camundongos (dosagem de duração de 91 semanas) com doses de até e, incluindo, 900 mg/kg/dia e 1.500 mg/kg/dia, respectivamente. Nenhuma evidência de carcinogenicidade com o cloridrato de metformina foi encontrada tanto em camundongos machos ou fêmeas. Do mesmo modo, não foi observado potencial tumorigênico com cloridrato de metformina, em ratos do sexo masculino. Houve, no entanto, um aumento da incidência de pólipos uterinos estromais benignos em ratos fêmeas tratadas com 900 mg/kg/dia. Em um segundo conjunto de estudos de carcinogenicidade realizado com uma forma de libertação lenta de cloridrato de metformina, a aplicação dérmica de metformina com camundongos transgênicos durante 26 semanas e com aplicação oral de metformina a ratos durante 104 semanas, em doses de até 1.200 mg/kg/dia não foram associados com evidência de carcinogenicidade.
Toxicidade reprodutiva e no desenvolvimento
A metformina não afetou a fertilidade ou mostrou ter efeitos teratogênicos em ratos com doses de até 600 mg/kg.