 Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)
Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)Qual a ação da substância do Praluent?
Resultados de Eficácia
Resumo do Programa de Estudos Clínicos de Fase 3 – esquema posológico de 75 mg e/ou 150 mg a cada 2 semanas (Q2W)
A eficácia do alirocumabe foi investigada em dez estudos clínicos fase 3 (cinco estudos controlados com placebo e cinco estudos controlados com ezetimiba), envolvendo a randomização de 5296 pacientes com hipercolesterolemia (não familiar e familiar heterozigótica) ou dislipidemia mista, com 3188 pacientes randomizados para alirocumabe. Três dos dez estudos foram conduzidos exclusivamente em pacientes com hipercolesterolemia familiar heterozigótica (HFHe). A maioria dos pacientes no programa de fase 3 estava já sendo tratada com estatina na dose máxima tolerada, associada ou não a outros hipolipemiantes, e apresentava risco cardiovascular (CV) alto ou muito alto. Dois estudos foram conduzidos exclusivamente com pacientes que não estavam sendo tratados concomitantemente com uma estatina, incluindo um estudo em pacientes com intolerância documentada à estatina.
Dois estudos (LONG TERM e HIGH FH), envolvendo um total de 2416 pacientes, foram realizados com a dose de 150 mg uma vez a cada duas semanas apenas. Oito estudos testaram a dose de 75 mg uma vez a cada duas semanas com titulação para 150 mg uma vez a cada duas semanas na semana 12 nos pacientes que não atingiram seus alvos predefinidos de LDL-C com base no seu risco CV na semana 8.
As características demográficas basais foram similares entre os braços alirocumabe e controle. A idade dos pacientes estudados variou de 18 a 89 anos (idade média de 60 anos); 38% eram mulheres; a maioria dos pacientes era caucasiana (90%), 5% eram negros, 2% asiáticos; o índice de massa corpórea (IMC) médio foi 30 kg/m2. Nos estudos de fase 3, 31% dos pacientes eram portadores de diabetes mellitus tipo 2 e 64% dos pacientes apresentavam histórico de doença coronariana.
O desfecho primário de eficácia em todos os estudos de fase 3 foi a porcentagem média de redução de LDL-C na semana 24 a partir do basal, em comparação ao placebo ou ezetimiba. Todos os estudos atingiram o desfecho primário.
Em geral, a administração de alirocumabe também resultou em uma maior redução percentual estatisticamente significativa do colesterol total, do colesterol não HDL, da Apo B e da Lp(a), em comparação ao placebo/ezetimiba, sendo os pacientes tratados ou não concomitantemente com uma estatina. O alirocumabe também reduziu os triglicérides, e aumentou HDL-C e Apo A-1 em comparação ao placebo. Para resultados detalhados ver as tabelas a seguir.
A redução de LDL-C foi verificada independentemente de idade, sexo, índice de massa corpórea (IMC), raça, níveis basais de LDL-C, e diagnóstico de HFHe, dislipidemia mista ou diabetes mellitus. A redução de LDL-C foi consistente independentemente do uso concomitante de estatinas em diferentes doses.
Uma proporção significativamente maior de pacientes atingiu níveis de LDL-C ˂ 70 mg/dL no grupo alirocumabe em comparação ao grupo placebo ou ezetimiba na semana 12 e semana 24.
Em estudos utilizando o regime de titulação progressiva baseada em critérios, a maioria dos pacientes alcançou o alvo predefinido de LDL-C (com base em seu nível de risco CV) com dosagem de 75 mg uma vez a cada duas semanas e a maioria dos pacientes mantiveram o tratamento nessa dosagem.
O efeito hipolipemiante do alirocumabe foi observado dentro de 15 dias após a primeira dose, alcançando efeito máximo em aproximadamente 4 semanas. A eficácia foi mantida ao longo da duração do tratamento do estudo (até 78 semanas no estudo LONG TERM).
Após a interrupção do alirocumabe, não se observou efeito rebote nos níveis de LDL-C, e os níveis de LDL-C retornaram gradualmente aos níveis basais.
As tabelas a seguir resumem a variação percentual média a partir do LDL-C basal com alirocumabe na semana 12 (antes da titulação) e na semana 24 (desfecho primário) com base em análises conjuntas de todos os estudos de fase 3.
Tabela 1 - Variação percentual média a partir do LDL-C basal com alirocumabe na semana 12 (antes da titulação) e na semana 24 (desfecho primário) em análise conjunta de 10 estudos de fase 3 controlados com placebo, em pacientes tratados com estatinas a
Semana 12 | ||
Dosagem | Alirocumabe (efeito aditivo além da estatina) | Placebo (efeito aditivo além da estatina) |
75 mg Q2W | -44,5% | +4,1% |
150 mg Q2W | -62,6% | +1,1% |
Semana 24 | ||
Dosagem | Alirocumabe (efeito aditivo além da estatina) | Placebo (efeito aditivo além da estatina) |
75/150 mg Q2W (estudos de titulação)b | -48,6% | +4,2% |
150 mg Q2W | -60,4% | +0,5% |
a Baseado em análises ITT – população com intenção de tratar. Inclui todos os dados de lípides durante toda a duração do estudo, independente da aderência ao tratamento do estudo.
b A dosagem foi titulada para 150 mg uma vez a cada duas semanas em 228 (34,5%) pacientes a partir da semana 12 de tratamento.
O LDL-C basal médio na análise conjunta de estudos de titulação (COMBO I, FH I e FH II) foi 129,1 mg/dL no grupo alirocumabe e 129,9 md/dL no grupo placebo.
O LDL-C basal médio na análise conjunta de estudos utilizando a dose de 150 mg uma vez a cada duas semanas (LONG TERM, HIGH FH) foi 126,0 mg/dL no grupo alirocumabe e 125,4 mg/dL no grupo placebo.
Os resultados individuais para cada um dos estudos são apresentados na Figura 1.
Tabela 2 - Variação percentual média a partir do LDL-C basal com alirocumabe na semana 12 (antes da titulação) e na semana 24 (desfecho primário) na análise conjunta dos estudos de fase 3 controlados com ezetimibaa
| - | Sem tratamento concomitante com estatina | Com tratamento concomitante com estatina | ||
Semana 12 | ||||
Dosagem | Alirocumabe | Ezetimiba | Alirocumabe (efeito aditivo além da estatina) | Ezetimiba (efeito aditivo além da estatina) |
75 mg Q2W | -47,4% | -16,7% | -49,2% | -22,3% |
Semana 24 | ||||
Dosagem | Alirocumabe | Ezetimiba | Alirocumabe (efeito aditivo além da estatina) | Ezetimiba (efeito aditivo além da estatina) |
75/150 mg Q2W (estudos de titulação)b | -45,6% | -14,8% | -48,9% | -19,3% |
a Baseado em análises ITT – população com intenção de tratar, inclui todos os dados de lípides durante toda a duração do estudo, independente da aderência ao tratamento do estudo.
b A dosagem foi titulada para 150 mg uma vez a cada duas semanas em 180 (22,9%) pacientes a partir da semana 12 de tratamento.
O LDL-C basal médio nas análises conjuntas de estudos sem tratamento com estatina (mono alternative) foi 176,5 mg/dL e 177,7 mg/dL no grupo ezetimiba.
O LDL-C basal médio nas análises conjuntas de estudos com tratamento com estatina (COMBO II, OPTIONS I e II) foi 109,3 mg/dL no grupo alirocumabe e 105,0 mg/dL no grupo ezetimiba.
Os resultados individuais para cada um dos estudos estão incluídos abaixo.
Nas análises conjuntas de estudos fase 3 que permitiram a titulação, dentre o subgrupo de pacientes titulados, um aumento de 75 mg uma vez a cada duas semanas para 150 mg uma vez a cada duas semanas de alirocumabe na semana 12 resultou em uma redução adicional média de 14% no LDL-C em pacientes em tratamento concomitante com estatina. Em pacientes não tratados com estatina, a titulação de alirocumabe resultou em uma redução adicional média de 3% no LDL-C, sendo a maioria dos efeitos observados em aproximadamente 25% dos pacientes que atingiram pelo menos 10% de redução adicional no LDL-C após a titulação. Pacientes titulados para 150 mg uma vez a cada duas semanas apresentavam um LDL-C basal médio mais elevado.
A Figura 1 apresenta a redução média no LDL-C basal obtida com alirocumabe na semana 12 (antes da titulação) em cada um dos estudos de fase 3. Essa figura demonstra a eficácia das dosagens de 75 mg uma vez a cada duas semanas e 150 mg uma vez a cada duas semanas. Os resultados da semana 24 estão fornecidos na descrição dos estudos individuais.
Figura 1 - Sinopse da redução percentual média do LDL-C basal obtida com alirocumabe na semana 12 (antes da titulação) em cada um dos estudos de fase 3.
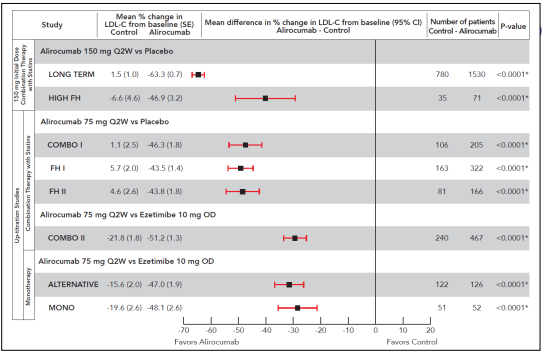
Terapia combinada com estatina
Estudos de fase 3 controlados com placebo em pacientes com hipercolesterolemia primária ou dislipidemia mista em uso de estatina.
Estudo LONG TERM
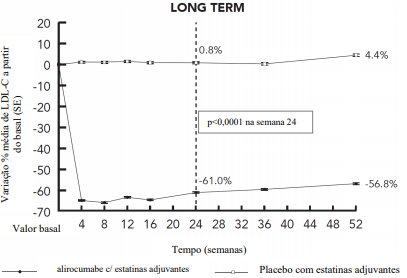
Este estudo multicêntrico, duplo-cego, controlado com placebo, de 18 meses de seguimento, incluiu 2310 pacientes (1530 pacientes no grupo alirocumabe e 780 pacientes no grupo placebo) com hipercolesterolemia primária com risco CV alto ou muito alto, e em uso de estatina na dose máxima tolerada, com ou sem associação de outros hipolipemiantes. Os pacientes receberam alirocumabe em uma dose de 150 mg uma vez a cada duas semanas ou placebo, em adição aos hipolipemiantes já em uso antes da inclusão no estudo. O estudo LONG TERM incluiu 17,7% de pacientes com HFHe, 34,6% com diabetes mellitus tipo 2, e 68,6% com histórico de doença arterial coronariana. A duração média de tratamento foi de 64,6 semanas, com a maioria de pacientes tratados por um mínimo de 52 semanas e 607 pacientes com acompanhamento de 18 meses. Na semana 24, a diferença média do tratamento em relação ao placebo na variação percentual do LDL-C basal foi de -61,9% (IC 95%: -64,3%, -59,4%; p ˂ 0,0001). Para resultados detalhados ver Tabela 3 e Figura 2. 82,1% dos pacientes no grupo alirocumabe atingiram um LDL-C < 70 mg/dL na semana 12, comparado a 7,2% dos pacientes no grupo placebo. A redução no LDL-C foi verificada independentemente de idade, gênero, índice de massa corpórea (IMC), raça e níveis basais de LDL-C. Os resultados de eficácia foram consistentes tanto em pacientes com HFHe como naqueles sem HFHe, incluindo pacientes com dislipidemia mista e portadores de diabetes mellitus. A redução do LDL-C foi consistente independentemente do uso concomitante de estatinas e de suas dosagens.
Tabela 3 - Variação percentual média no LDL-C a partir do basala em pacientes com hipercolesterolemia primária tratados com alirocumabe ou placebo (ambos em uso de estatina na dose máxima tolerada) – Análise ITTb
Estudo LONG TERM | ||||||||||
Grupo de tratamento | N | LDL-C | LDL-Cc | Colesterol Total | Não HDL-C | Apo B | Lp(a) | TG | HDL -C | Apo A-1 |
Semana 12 | ||||||||||
Placebo (com estatina adjuvante) | 780 | 1,5 | 1,4 | 0,2 | 0,9 | 0,5 | -3,1 | 1,2 | 0,2 | 0.6 |
Alirocumabe (150 mg) (com estatina adjuvante) | 1530 | -63,3 | -64,2 | -38,8 | -53,7 | -55,5 | -28,2 | -16,7 | 5,8 | 4,6 |
Semana 24 | ||||||||||
Placebo (com estatina adjuvante) | 780 | 0,8 | 0,7 | -0,3 | 0,7 | 1,2 | -3,7 | 1,8 | -0,6 | 1,2 |
Alirocumabe (150 mg) (com estatina adjuvante) | 1530 | 0 -61,0d | -62,8 | -37,8 | -51,6 | -52,8 | -29,3 | -15,6 | 4,0 | 4,0 |
a Valor basal – em tratamento com estatina com ou sem outro hipolipemiante. O LDL-C basal médio foi 122,7 mg/dL no grupo alirocumabe e 121,9 mg/dL no grupo placebo.
b Análise ITT – população intenção de tratar, inclui todos os dados de lípides durante toda a duração do estudo, independentemente da aderência ao tratamento do estudo.
c Análise em tratamento – análise restrita ao período de tempo em que os pacientes realmente receberam o tratamento.
d Este corresponde à variação absoluta média a partir do basal de -74,5 mg/dL.
A diferença vs. placebo foi estatisticamente significativa na semana 24 para todos os lípides/lipoproteínas listados na tabela.
Figura 2 - LDL-C ao longo do tempo: Variação percentual média a partir do LDL-C basal até 52 semanas de tratamento - Estudo LONG TERM – (análise ITT)
Estudo COMBO I
O estudo COMBO I foi um estudo multicêntrico, duplo-cego, controlado com placebo, de 52 semanas de acompanhamento, que incluiu 311 pacientes (205 no grupo alirocumabe e 106 pacientes no grupo placebo) categorizados como de risco CV muito alto e fora do seu alvo de LDL-C pré-definido, em uso de estatina na dose máxima tolerada, com ou sem outro hipolipemiante associado. Os pacientes receberam 75 mg de alirocumabe uma vez a cada duas semanas ou placebo em adição à terapia hipolipemiante já instaurada. Na semana 12 procedeu-se paratitulação de dose de alirocumabe para 150 mg uma vez a cada duas semanas nos pacientes com LDL-C ≥ 70 mg/dL. O LDL-C médio basal era 100,2 mg/dL no grupo alirocumabe e 106,0 mg/dL no grupo placebo. A variação percentual média de LDL-C a partir do basal com alirocumabe (análise ITT) foi -46,3% na semana 12 e -48,2% na semana 24 comparada a 1,1% na semana 12 e -2,3% na semana 24 para o placebo. Isto correspondeu a uma variação absoluta média a partir do basal na semana 24 de -50,3% mg/dL.
Na semana 24, a diferença média do tratamento em relação ao placebo na variação percentual dos níveis de LDL-C a partir do basal foi de -45,9% (IC 95%: - 52,5%, -39,3%; p ˂ 0,0001). Na semana 12 (antes da titulação), 76,0% dos pacientes no grupo alirocumabe atingiram um nível de LDL-C de < 70 mg/dL em comparação a 11,3% no grupo placebo. A dosagem foi titulada para 150 mg uma vez a cada duas semanas em 32 (16,8%) dos pacientes tratados além de 12 semanas. No subgrupo de pacientes titulados na semana 12, uma redução adicional média de 22,8% no nível de LDL-C foi atingida na semana 24. A diferença em relação ao placebo foi estatisticamente significativa na semana 24 para todos os parâmetros lipídicos avaliados, exceto para TG e Apo A-1.
Estudos fase 3 controlados com placebo (com estatina adjuvante) em pacientes com hipercolesterolemia familiar heterozigótica (HFHe)
Estudos FH I e FH II
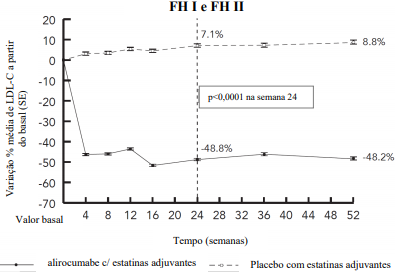
Dois estudos multicêntricos, controlados com placebo, duplo-cegos, de 18 meses de duração, incluíram 732 pacientes (488 no grupo alirocumabe e 244 pacientes no grupo placebo, com a maioria de pacientes tratados por um mínimo de 52 semanas) com HFHe em uso de estatina na dose máxima tolerada, com ou sem outro hipolipemiante. Os pacientes receberam alirocumabe 75 mg uma vez a cada duas semanas ou placebo em adição à medicação hipolipemiante já em uso. Nos pacientes com LDL-C ≥ 70 mg/dL ocorreu titulação da dose de alirocumabe para 150 mg uma vez a cada duas semanas na semana 12. Na semana 24, a diferença média do tratamento em relação ao placebo na variação percentual dos níveis de LDL-C a partir do basal foi -55,8% (IC 95%: -60,0%, -51,6%; p ˂ 0,0001). Para resultados detalhados ver Tabela 4 e Figura 3. Na semana 12 (antes da titulação), 50,2% dos pacientes atingiram um nível de LDL-C < 70 mg/dL em comparação a 0,6% no grupo placebo. No subgrupo de pacientes titulados na semana 12, uma de redução adicional média de 15,7% nos níveis de LDL-C foi alcançada na semana 24.
Tabela 4 - Variação percentual média no LDL-C a partir do basala em pacientes com hipercolesterolemia familiar heterozigótica tratados com alirocumabe ou placebo (ambos em terapia com estatina na dose máxima tolerada) – Análise ITTb
Análise Conjunta dos Estudos FH I e FH II | ||||||||||
Grupo de tratamento | N | LDL-C | LDL-Cc | Colesterol Total | Não HDL-C | Apo B | Lp(a) | TG | HDL-C | Apo A-1 |
Semana 12 | ||||||||||
Placebo (com estatina adjuvante) | 244 | 5,4 | 5,3 | 3,9 | 4,9 | 1,8 | -4,6 | 1,2 | 2,1 | -0,6 |
Alirocumabe (75 mg) (com estatina adjuvante) | 488 | -43,6 | -44,0 | -27,7 | -38,2 | -34,8 | -22,6 | -8,0 | 6,3 | 2,0 |
Semana 24 | ||||||||||
Placebo (com estatina adjuvante) | 244 | 7,1 | 6,8 | 5,5 | 7,4 | 1,9 | -8,5 | 4,3 | 0,2 | -0,4 |
Alirocumabe (75/150 mg) (com estatina adjuvante) e | 488 | -48,8d | -49,3 | -31,2 | -42,8 | -41,7 | -26,9 | -9,8 | 7,8 | 4,2 |
a Valor basal – com uma estatina isolada com ou sem outro hipolipemiante. Valor basal médio de LDL-C era 141,3 mg/dL no grupo alirocumabe e 140,9 mg/dL no grupo placebo.
b Análise ITT – população intenção de tratar, inclui todos os dados de lípides durante toda a duração do estudo independente da aderência ao tratamento.
c Análise em tratamento – análise restrita ao período de tempo em que os pacientes realmente receberam o tratamento.
d Este corresponde a variação absoluta média a partir do basal de -71,1 mg/dL.
e A dosagem foi titulada para 150 mg uma vez a cada duas semanas em 196 (41,8%) pacientes tratados além de 12 semanas.
A diferença vs placebo foi estatisticamente significativa na semana 24 para todos os lípides/lipoproteínas listadas na tabela.
Figura 3 - LDL-C ao longo do tempo: variação percentual média a partir do basal até 52 semanas – Análise Conjunta dos Estudos FH (FH I e FH II) (Análise ITT)
Estudo HIGH FH
Um terceiro estudo multicêntrico, duplo-cego, controlado com placebo, com duração de 18 meses, incluiu 106 pacientes HFHe (71 pacientes no grupo alirocumabe e 35 pacientes no grupo placebo, com uma maioria de pacientes tratados por um mínimo de 52 semanas) em uso de estatina na dose máxima tolerada, com ou sem outro hipolipemiante, e LDL-C basal ≥ 160 mg/dL. Os pacientes receberam alirocumabe na dose de 150 mg uma vez a cada duas semanas ou placebo em adição à medicação hipolipemiante já em uso. O LDL-C médio basal era 196,3 mg/dL no grupo alirocumabe e 201,0 mg/dL no grupo placebo. A variação percentual média de LDL-C a partir do basal com alirocumabe (análise ITT) foi -46,9% na semana 12 e -45,7% na semana 24 comparado a -6,6% na semana 12 e -6,6% na semana 24 para o placebo. Isto correspondeu a uma variação absoluta média a partir do basal de -90,8 mg/dL na semana 24. Na semana 24, a diferença média do tratamento em relação ao placebo na variação percentual dos níveis de LDL-C a partir do basal foi -39,1% (IC 95%: -51,1%, -27,1%; p ˂ 0,0001). Variações médias para todos os outros lípides/lipoproteínas foram similares aos estudos FH I e FH II, entretanto, não se alcançou significância estatística para TG, HDL-C e Apo A-1.
Estudo fase 3 controlado com ezetimiba (com estatina adjuvante) em pacientes com hipercolesterolemia primária ou dislipidemia mista.
Estudo COMBO II
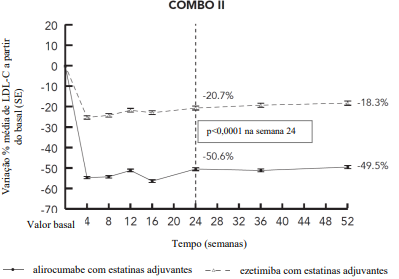
Um estudo multicêntrico, duplo-cego, controlado com ezetimiba, de 2 anos de duração, incluiu 707 pacientes (467 pacientes no grupo alirocumabe e 240 pacientes no grupo ezetimiba, com a maioria dos pacientes tratados por um mínimo de 52 semanas) categorizados como de risco CV muito alto e fora do seu alvo pré-definido de LDL-C e em uso da estatina na dose máxima tolerada. Os pacientes receberam 75 mg uma vez a cada duas semanas de alirocumabe ou ezetimiba 10 mg uma vez ao dia em adição ao tratamento com estatinas. Titulação de dosagem de alirocumabe para 150 mg uma vez a cada duas semanas ocorreu na semana 12 em pacientes com LDL-C ≥ 70 mg/dL. Na semana 24, a diferença média do alirocumabe em relação à ezetimiba na variação percentual do nível de LDL-C a partir do basal foi - 29,8% (IC 95%: -34,4%, -25,3%; p ˂ 0,0001). Para resultados detalhados ver Tabela 5 e Figura 4. Na semana 12 (antes da titulação), 77,2% dos pacientes atingiram um LDL-C de < 70 mg/dL em comparação a 46,2% no grupo ezetimiba. Dentre o subgrupo de pacientes titulados na semana 12, uma redução adicional média de 10,5% no nível de LDL-C foi alcançada na semana 24.
Tabela 5 - Variação percentual média de LDL-C a partir do basala em pacientes com hipercolesterolemia primária tratados com alirocumabe ou ezetimiba (ambos em uso de estatina na dose máxima tolerada) – Análise ITTb
Estudo COMBO II | ||||||||||
Grupo de tratamento | N | LDL-C | LDL-Cc | Colesterol Total | Não HDL-C | Apo B | Lp(a) | TG | HDL-C | Apo A1 |
Semana 12 | ||||||||||
Ezetimiba (10 mg) (com estatina adjuvante) | 240 | -21,8 | -22,7 | -15,1 | -20,6 | -17,2 | 1,1 | -15,3 | 2,8 | -2,9 |
Alirocumabe (75 mg) (com estatina adjuvante) | 467 | -51,2 | -52,4 | -29,4 | -42,6 | -39,7 | -22,1 | -13,5 | 8,7 | 1,5 |
Semana 24 | ||||||||||
Ezetimiba (10 mg) (com estatina adjuvante) | 240 | -20,7 | -21,8 | -14,6 | -19,2 | -18,3 | -6,1 | -12,8 | 0,5 | -1,3 |
Alirocumabe (75/150 mg)e (com estatina adjuvante) | 467 | -50,6d | -52,4 | -29,3 | -42,1 | -40,7 | -27,8 | -13 | 8,6 | 5,0 |
a Valor basal – em estatina isolada. LDL-C médio basal era 108,6 mg/dL no grupo alirocumabe e 104,6 mg/dL no grupo ezetimiba.
b Análise ITT – população intenção de tratar, inclui todos os dados de lípides por toda duração do estudo independente da aderência ao tratamento do estudo.
c Análise em tratamento – análise restrita ao período de tempo que os pacientes realmente receberam o tratamento.
d Este corresponde a variação absoluta média a partir do basal de LDL-C de -55,4 mg/dL na semana 24.
e Dosagem foi titulada para 150 mg uma vez a cada duas semanas em 82 (18,4%) pacientes tratados além de 12 semanas.
A diferença vs ezetimiba foi estatisticamente significativa na semana 24 para todos os lípides/lipoproteínas exceto para TG, e Apo A-1.
Figura 4 - LDL-C ao longo do tempo: variação percentual média a partir do basal até 52 semanas – estudo COMBO II (análise ITT)
Monoterapia ou como complemento à terapia hipolipemiante sem estatina Estudos fase 3 controlado com ezetimiba em pacientes com hipercolesterolemia primária (sem estatina adjuvante)
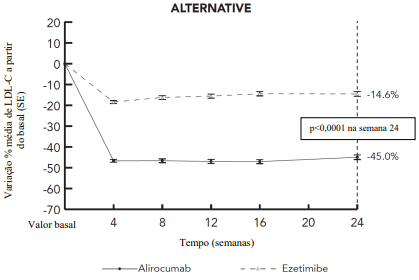
Estudo ALTERNATIVE
Este estudo multicêntrico, duplo-cego, controlado com ezetimiba de 24 semanas incluiu 248 pacientes (126 no grupo alirocumabe e 122 no grupo ezetimiba) com intolerância documentada à estatina devido aos sintomas relacionados aos musculoesqueléticos. Pacientes receberam alirocumabe 75 mg uma vez a cada duas semanas ou ezetimiba 10 mg uma vez ao dia, ou atorvastatina 20 mg uma vez ao dia (como um braço de novo desafio). A titulação da dosagem de alirocumabe para 150 mg uma vez a cada duas semanas ocorreu na semana 12 em pacientes com LDL-C ≥ 70 mg/dL ou ≥ 100 mg/dL, dependendo do seu nível de risco CV. Na semana 24, a diferença média do tratamento em relação à ezetimiba na variação percentual de LDL-C a partir do basal foi de -30,4% (IC 95%: -36,6%, -24,2%; p ˂ 0,0001). Para resultados detalhados ver Tabela 6 e Figura 5. Na semana 12 (antes da titulação), 34,9% dos pacientes atingiram um LDL-C de < 70 mg/dL em comparação a 0% no grupo ezetimiba. Dentre o subgrupo de pacientes titulados na semana 12, uma redução média adicional de 3,6% no LDL-C foi alcançada na semana 24.
O estudo avaliou pacientes que não toleravam pelo menos duas estatinas (pelo menos uma na dosagem mais baixa aprovada) e recrutou apenas pacientes dispostos a ser tratados novamente com uma estatina. O braço de novo desafio com a estatina foi incluído para validar posteriormente o diagnóstico da intolerância a estatina de forma cega. Nesses pacientes com histórico de intolerância a estatina, eventos adversos musculoesqueléticos ocorreram a uma taxa menor no grupo alirocumabe (32,5%) em comparação ao grupo da atorvastatina (46,0%) (HR=0,61 [IC 95%: 0,38, 0,99]), e uma menor porcentagem de pacientes no grupo alirocumabe (15,9%) descontinuou o tratamento do estudo devido aos eventos adversos musculoesqueléticos em comparação ao grupo da atorvastatina (22,2%). Essas taxas de descontinuação devido aos eventos adversos musculoesqueléticos no estudo ALTERNATIVE foram maiores do que em outros estudos fase 3. Nos cinco estudos controlados com placebo em pacientes em uso de estatina na dose máxima tolerada (n=3752), a taxa de descontinuação devido aos eventos adversos musculoesqueléticos foi 0,4% no grupo alirocumabe e 0,5% no grupo placebo.
Tabela 6 - Variação percentual média de LDL-C a partir do basala em pacientes com hipercolesterolemia primária tratados com alirocumabe ou ezetimiba (sem estatina adjuvante) – Análise ITTb
Estudo ALTERNATIVE | ||||||||||
Grupo de tratamento | N | LDL-C | LDL-Cc | Colesterol Total | Não HDL-C | Apo B | Lp(a) | TG | HDL-C | Apo A1 |
Semana 12 | ||||||||||
Ezetimiba (10 mg) | 122 | -15,6 | -18,0 | -11,6 | -15,8 | -11,6 | -4,5 | -9,4 | 7,6 | 3,9 |
Alirocumabe (75 mg) | 126 | -47,0 | -51,2 | -32,7 | -41,5 | -36,1 | -21,7 | -8,0 | 9,0 | 5,5 |
Semana 24 | ||||||||||
Ezetimiba (10 mg) | 122 | -14,6 | -17,1 | -10,9 | -14,6 | -11,2 | -7,3 | -3,6 | 6,8 | 2,9 |
Alirocumabe (75/150 mg) e | 126 | -45,0d | -52,2 | -31,8 | -40,2 | -36,3 | -25,9 | -9,3 | 7,7 | 4,8 |
a Valor basal – em estatina isolada. LDL-C médio basal era 191,1 mg/dL no grupo alirocumabe e 193,5 mg/dL no grupo ezetimiba.
b Análise ITT – população intenção de tratar, inclui todos os dados de lípides por toda duração dos estudos independente da aderência ao tratamento do estudo.
c Análise em tratamento – análise restrita ao período de tempo que os pacientes realmente receberam o tratamento.
d Este corresponde a variação absoluta média de -84,2 mg/dL na semana 24.
e A dosagem foi titulada para 150 mg uma vez a cada duas semanas em 54 (49,5%) pacientes tratados além de 12 semanas.
A diferença vs ezetimiba foi estatisticamente significativa na semana 24 para LDL-C, Colesterol total, Não HDL-C, Apo B, e Lp(a).
Figura 5 - LDL-C ao longo do tempo: variação percentual média a partir do basal até 24 semanas – estudo ALTERNATIVE (análise ITT)
Estudo MONO
Este estudo multicêntrico, duplo-cego, controlado com ezetimiba de 24 semanas incluiu 103 pacientes (52 pacientes no grupo alirocumabe e 51 pacientes no grupo ezetimiba) com risco CV moderado, sem uso de estatinas ou outros hipolipemiantes, e LDL-C basal entre 100 mg/dL e 190 mg/dL. Os pacientes receberam alirocumabe 75 mg uma vez a cada duas semanas ou ezetimiba 10 mg uma vez ao dia. A titulação de dosagem de alirocumabe para 150 mg uma vez a cada duas semanas ocorreu na semana 12 em pacientes com LDL-C ≥ 70 mg/dL. O LDL-C basal era 141,1 mg/dL no grupo alirocumabe e 138,3 mg/dL no grupo ezetimiba. A variação percentual média de LDL-C a partir do basal com alirocumabe (análise ITT) foi -48,1% na semana 12 e -47,2% na semana 24 comparada a -19,6% na semana 12 e - 15,6% na semana 24 para ezetimiba. Isto corresponde a uma variação absoluta média a partir do basal na semana 24 de -66,9 mg/dL. Na semana 24, a diferença média entre o tratamento e ezetimiba na variação percentual do nível de LDL-C a partir do basal foi -31,6% (IC 95%: -40,2%, -23,0%; p ˂ 0,0001). Na semana 12 (antes da titulação), 57,7% dos pacientes atingiram um LDL-C de < 70 mg/dL em comparação a 0% no grupo ezetimiba. A dosagem foi titulada para 150 mg em 14 (30,4%) pacientes tratados além de 12 semanas. Dentre o subgrupo de pacientes titulados na semana 12, uma redução adicional média de 1,4% em LDL-C foi alcançado na semana 24. A diferença vs ezetimiba foi estatisticamente significativa na semana 24 para LDL-C, Colesterol total, Colesterol não HDL e Apo B.
Outros estudos
Estudos OPTIONS I e OPTIONS II
Dois estudos adicionais multicêntricos, duplo-cegos, controlados ativamente de 24 semanas foram realizados em 643 pacientes (combinados) com hipercolesterolemia primária, risco CV alto ou muito alto não adequadamente controlados com atorvastatina em dose moderada (20 mg ou 40 mg, OPTIONS I) ou rosuvastatina (10 mg ou 20 mg, OPTIONS II). A titulação da dosagem do alirocumabe de 75 mg uma vez a cada duas semanas para 150 mg uma vez a cada duas semanas ocorreu na semana 12 nos pacientes com LDL-C ≥ 70 mg/dL ou 100 mg/dL, dependendo do nível de risco CV. Os níveis médios basais de LDL-C variaram de 105,1 mg/dL (OPTIONS I) a 111,3 mg/dL (OPTIONS II). Na semana 24, a variação percentual média de LDL-C a partir do basal com alirocumabe em adição a doses moderadas de atorvastatina foi de -48,5% no OPTIONS I, e -42,7% quando adicionado a doses moderadas de rosuvastatina no OPTIONS II.
Esquema posológico a cada 4 semanas (Q4W)
Estudo CHOICE I
Um estudo multicêntrico, duplo-cego, placebo-controlado, de 48 semanas e com 540 paciente na dose máxima tolerada de uma estatina, com ou sem outra terapia modificadora de lipídeo (308 no grupo de alirocumabe 300 mg a cada 4 semanas (Q4W), 76 no grupo de alirocumabe 75 mg a cada duas semanas (Q2W) e 156 no grupo placebo), e 252 pacientes não tratados com uma estatina (144 no grupo alirocumabe 300 mg Q4W, 37 no grupo alirocumabe 75 mg Q2W e 71 no grupo placebo). Os pacientes receberam ou alirocumabe 300 mg Q4W, alirocumabe 75 mg Q2W ou placebo em adição a sua terapia modificadora de lipídeo existente (estatina, terapia sem estatina ou somente dieta). No geral, 71,6% dos pacientes foram classificados com risco cardiovascular (CV) alto ou muito alto e fora do LDL-C alvo. Nos braços com alirocumabe, o ajuste de dose para 150 mg a cada duas semanas nos pacientes com LDLC ≥ 70 mg/dL ou ≥ 100 mg/dL ocorreu na semana 12, dependendo de seus níveis de risco cardiovascular (CV) ou nos pacientes que não tiveram uma redução de pelo menos 30% do LDL-C a partir do da linha de base.
No grupo de pacientes com histórico de estatina, o valor médio inicial de LDL-C foi 112,7 mg/dL. Na semana 12, a mudança percentual média no LDL-C (análise ITT), a partir do basal, com alirocumabe 300 mg a cada 4 semanas foi -55,3% comparado a +1,1% para o placebo. Na semana 12 (antes do ajuste de dose), 77,3% dos pacientes tratados com alirocumabe 300 mg a cada 4 semanas atingiram um LDL-C de ˂ 70 mg/dL em comparação com 9,3% no grupo placebo. Na semana 24, a mudança percentual média no LDL-C (análise ITT), a partir do basal, para alirocumabe 300 mg a cada 4 semanas/150 mg a cada duas semanas foi -58,8% em comparação com -0,1% para o placebo. Na semana 24, a diferença de tratamento média em termos de mudança percentual no LDL-C, a partir do basal, para o alirocumabe 300 mg a cada quatro semanas/150 mg a cada duas semanas em relação ao placebo foi -58,7 % (IC 97,5%: -65,0%, - 52,4%; p-value: ˂ 0,0001). Nos pacientes tratados além de 12 semanas, a dose foi ajustada para 150 mg a cada duas semanas em 56 (19,3%) dos 290 pacientes no braço com alirocumabe 300 mg a cada 4 semanas. Entre o subgrupo de pacientes que tiveram a dose ajustada para 150 mg a cada duas semanas na semana 12, uma redução adicional 25,4% no LDL-C foi alcançada na semana 24.
No grupo de pacientes que não estavam recebendo um tratamento concomitante com estatina, o valor médio inicial de LDL-C foi 142,1 mg/dL. Na semana 12, a mudança percentual no LDL-C (análise ITT), a partir do basal, com alirocumabe 300 mg a cada 4 semanas foi -58,4% comparado com +0,3% para o placebo. Na semana 12 (antes do ajuste de dose), 65,2% dos pacientes tratados com alirocumabe 300 mg a cada 4 semanas atingiram um LDL-C de ˂ 70 mg/dL em comparação com 2,8% no grupo placebo. Na semana 24, a mudança percentual media no LDL-C (análise ITT), a partir do basal, para alirocumabe 300 mg a cada 4 semanas/150 mg a cada duas semanas foi -52,7% comparado com -0,3% para o placebo. Na semana 24, a diferença de tratamento média em termos de mudança percentual no LDL-C, a partir do basal, para o alirocumabe 300 mg a cada quatro semanas/150 mg a cada duas semanas em relação ao placebo foi -52,4% (IC 97,5%: -59,8%, -45,0%; p-value: ˂ 0,0001). Nos pacientes tratados além de 12 semanas, a dose foi ajustada para 150 mg a cada duas semanas em 19 (14,7%) de 129 pacientes no braço com alirocumabe 300 mg a cada 4 semanas. Entre o subgrupo de pacientes que tiveram a dose ajustada para 150 mg a cada duas semanas na semana 12, uma redução adicional média de 7,3% no LDL-C foi alcançada na semana 24.
Em ambos os grupos, a diferença versus o placebo foi estatisticamente significante, na semana 24, para todos os parâmetros lipídicos, exceto para a Apo A-1 no subgrupo dos pacientes com histórico de estatina.
Estudo ODYSSEY OUTCOMES
Um estudo multicêntrico, duplo-cego, controlado por placebo em 18924 pacientes adultos (9462 alirocumabe; 9462 placebo) seguido por até 5 anos. Os pacientes apresentaram um evento de síndrome coronariana aguda (SCA) 4 a 52 semanas antes da randomização e foram tratados em regime de outros hipolipemiantes intensivo em estatina (definido como atorvastatina 40 ou 80 mg, ou rosuvastatina 20 ou 40 mg) ou na dose máxima tolerada dessas estatinas, com ou sem outro hipolipemiante. Todos os pacientes foram randomizados 1:1 para receber 75 mg de alirocumabe uma vez a cada duas semanas ou placebo uma vez a cada duas semanas. No mês 2, se uma diminuição adicional de LDL-C fosse necessária, com base nos critérios pré-especificados de LDL-C (LDL-C ≥ 50 mg/dL ou 1,29 mmol/L), o alirocumabe seria ajustado para 150 mg uma vez a cada duas semanas.
Para pacientes que tiveram sua dose ajustada para 150 mg uma vez a cada duas semanas e que tiveram dois valores consecutivos de LDL-C abaixo de 25 mg/dL (0,65 mmol/L), foi realizada uma redução da titulação de 150 mg uma vez a cada duas semanas para 75 mg uma vez a cada duas semanas. Pacientes com 75 mg uma vez a cada duas semanas que tiveram dois valores consecutivos de LDL-C abaixo de 15 mg/dL (0,39 mmol/L) foram trocados para placebo de forma cega. Aproximadamente 2615 (27,7%) de 9451 pacientes tratados com alirocumabe precisaram de ajuste de dose para 150 mg uma vez a cada duas semanas. Destes 2615 pacientes, 805 (30,8%) foram reduzidos para 75 mg uma vez a cada duas semanas. No total, 730 (7,7%) dos 9451 pacientes mudaram para placebo. A dose de 300 mg (Q4W) não foi avaliada neste estudo. Para efeitos de redução dos lipídios da dose de 300 mg (Q4W), consulte os resultados do estudo CHOICE I. Um total de 99,5% dos pacientes foram acompanhados vivos até o final do estudo. A duração média do acompanhamento foi de 33 meses.
O evento index de síndrome coronariana aguda (SCA) foi infarto do miocárdio em 83,2% dos pacientes (34,6% infarto do miocárdio com elevação do segmento ST (STEMI), 48,6% infarto do miocárdio sem elevação do segmento ST (NSTEMI)) e episódio de angina instável em 16,8% dos pacientes. Antes do indicador de evento de SCA, 19,2% tiveram infarto do miocárdio e 22,7% realizaram procedimentos de revascularização coronária (cirurgia de revascularização miocárdica (CRM)/(intervenção coronariana percutânea (ICP)). A maioria dos pacientes (88,8%) estava recebendo terapia com estatina de alta intensidade com ou sem outros hipolipemiantes na randomização. O valor médio de LDL-C basal foi de 92,4 mg/dL (2,39 mmol/L).
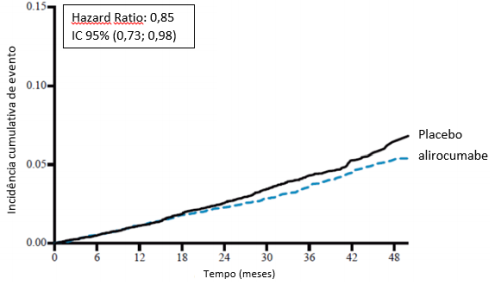
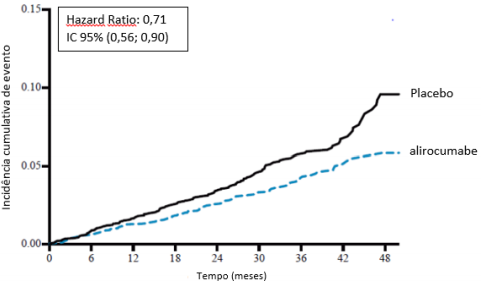
O alirocumabe reduziu significativamente o risco do desfecho composto primário da primeira ocorrência de eventos adversos cardiovasculares importantes (EACI) consistindo em morte por doença arterial coronariana (DAC), infarto do miocárdio (IAM) não fatal, acidente vascular cerebral isquêmico fatal e não fatal ou angina instável (AI) requerendo hospitalização (HR 0,85, IC 95%: 0,78, 0,93; p = 0,0003). O alirocumabe também reduziu significativamente os seguintes desfechos compostos: o risco de evento por DAC; evento maior por DAC; evento cardiovascular; e o conjunto de mortalidade por todas as causas, IAM não fatal e acidente vascular cerebral isquêmico não fatal. Uma redução da mortalidade por todas as causas também foi observada. No subgrupo de pacientes de alto risco com valores iniciais de LDL-C ≥ 100 mg/dL (≥ 2,59 mmol/L), os desfechos primários e secundários foram melhorados com o tratamento com alirocumabe, incluindo morte por DAC, morte CV e mortalidade por todas as causas. Os resultados são apresentados na Tabela 7 e Tabela 8.
O efeito do alirocumabe na redução do desfecho primário (MACE) foi observado no estudo ODYSSEY OUTCOMES após aproximadamente 1 ano de tratamento, consistente com o que foi observado nos ensaios de desfechos cardiovasculares realizados com outras terapias modificadoras de lipídios (especificamente estatinas). Para o subgrupo de pacientes com LDL-C basal ≥ 100 mg/dL (2.59 mmol/L), o efeito na redução de MACE foi observado após 6 meses. O efeito sobre a redução do LDL-C foi observado a partir do mês 1, que foi o primeiro ponto de tempo medido no estudo.
Tabela 7 - Eficácia do alirocumabe em ODYSSEY OUTCOMES (população geral)
Desfecho | Número de Eventos | Hazard Ratio IC 95% Valor p | ||
Alirocumabe n=9462 n (%) | Placebo n=9462 n (%) | |||
Desfecho primário (EACI) | 903 (9,5%) | 1052 (11,1%) | 0,85 (0,78; 0,93) 0,0003 | 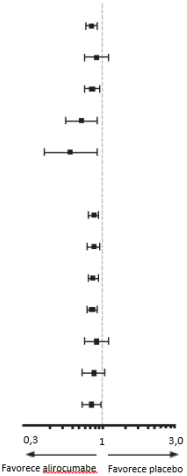 |
Morte por DAC | 205 (2,2%) | 222 (2,3%) | 0,92 (0,76; 1,11) 0,38 | |
IAM não fatal | 626 (6,6%) | 722 (7,6%) | 0,86 (0.77; 0,96) 0,006 | |
Acidente vascular cerebral isquêmico | 111 (1,2%) | 152 (1,6%) | 0.73 (0,57; 0,93) 0,01 | |
Angina instável | 37 (0,4%) | 60 (0,6%) | 0,61 (0,41; 0,92) 0,02 | |
Desfecho secundário | ||||
Evento por DAC | 1199 (12,7%) | 1349 (14,3%) | 0,88 (0.81; 0,95) 0,0013 | |
Evento maior por DAC | 793 (8,4%) | 899 (9,5%) | 0,88 (0,81; 0,94) 0,0003 | |
Evento cardiovascular | 1301 (13,7%) | 1474 (15,6%) | 0,87 (0,81; 0,94) 0,0003 | |
Mortalidade por todas as causas, IAM não fatal, acidente vascular cerebral isquêmico não fatal | 973 (10,3%) | 1126 (11,9%) | 0,86 (0,79; 0,93) 0,0003 | |
Morte por DAC | 205 (2,2%) | 222 (2,3%) | 0,92 (0,76; 1,11) 0,3824 | |
Morte cardiovascular | 240 (2,5%) | 271 (2,9%) | 0,88 (0,74; 1,05) 0,1528 | |
Todas as causas de mortalidade | 334 (3,5%) | 392 (4,1%) | 0,85 (0,73; 0,98) 0,0261 | |
a Angina instável que requer hospitalização.
b Evento por DAC definido como: evento maior por DAC, angina instável que requer hospitalização, procedimento de revascularização coronária por isquemia.
c Evento maior por DAC definido como: morte por DAC, IAM não fatal.
d Evento cardiovascular definido da seguinte forma: morte CV, qualquer evento coronariano não fatal e acidente vascular cerebral isquêmico não fatal.
e Não ajustado para comparações múltiplas.
Tabela 8 - Eficácia do alirocumabe no ODYSSEY OUTCOMES - LDL-C basal ≥ 100 mg/dL (≥ 2,59 mmol/L)
Desfecho | Número de Eventos | Hazard Ratio IC 95% Valor p | ||
| Alirocumabe n=2814 n (%) | Placebo n=2815 n (%) | |||
Desfecho primário (EACI) | 324 (11,5%) | 420 (14,9%) | 0,76 (0,65; 0,85) | 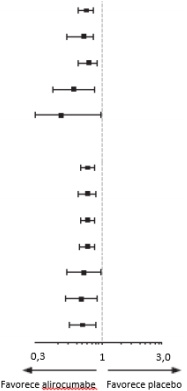 |
Morte por DAC | 69 (2,5%) | 96 (3,4%) | 0,72 (0.53; 0,98) | |
IAM não fatal | 231 (8,2%) | 290 (10,3%) | 0,79 (0,66; 0,93) | |
Acidente vascular cerebral isquêmico | 41 (1,5%) | 67 (2,4%) | 0,60 (0,41; 0,89) | |
Angina instável | 11 (0,4%) | 23 (0,8%) | 0,48 (0,23; 0,98) | |
Desfecho secundário | ||||
Evento por DAC | 404 (14,4%) | 508 (18%) | 0,78 (0,69; 0,89) | |
Evento maior por DAC | 285 (10,1%) | 364 (12,9%) | 0,77 (0,66; 0,90) | |
Evento cardiovascular | 441 (15,7%) | 552 (19,6%) | 0,78 (0,69; 0,89) | |
Mortalidade por todas as causas, IAM não fatal, acidente vascular cerebral isquêmico não fatal | 349 (12,4%) | 446 (15,8%) | 0,77 (0,67; 0,88) | |
Morte por DAC | 69 (2,5%) | 96 (3,4%) | 0,72 (0,53; 0,98) | |
Morte cardiovascular | 81 (2,9%) | 117 (4,2%) | 0,69 (0,52; 0,92) | |
Todas as causas de mortalidade | 114 (4,1%) | 161 (5,7%) | 0,71 (0,56; 0,90) | |
a Angina instável que requer hospitalização.
b Evento por DAC definido como: evento maior por DAC, angina instável que requer hospitalização, procedimento de revascularização coronária por isquemia.
c Evento por DAC principal definido como: morte por DAC, IAM não fatal.
d Evento cardiovascular definido da seguinte forma: morte CV, qualquer evento coronariano não fatal e acidente vascular cerebral isquêmico não fatal.
As estimativas de Kaplan-Meier da incidência cumulativa do desfecho primário e do desfecho de mortalidade por todas as causas para a população total de pacientes e para pacientes com LDL-C ≥ 100 mg/dL (≥ 2,59 mmol/L) ao longo do tempo são apresentadas nas Figuras 6 e 7.
Figura 6 - Incidência acumulada do ponto de extremidade composto primário ao longo de 4 anos em ODYSSEY OUTCOMES
População geral
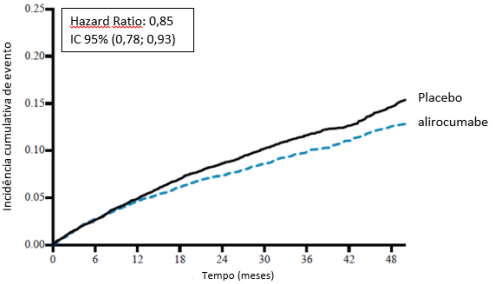
LDL-C basal ≥ 100 mg/dL
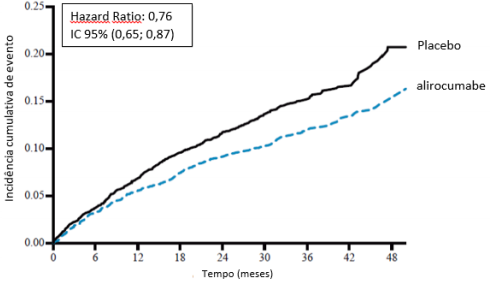
Figura 7 - Incidência acumulada de todas as causas de mortalidade ao longo de 4 anos em ODYSSEY OUTCOMES
População geral
LDL-C basal ≥ 100 mg/dL
Características Farmacológicas
Mecanismo de ação
O alirocumabe é um anticorpo monoclonal totalmente humano que se liga com alta afinidade e especificidade à pró-proteína convertase subtilisina/quexina tipo 9 (PCSK9).
A PCSK9 se liga aos receptores de lipoproteína de baixa densidade (LDLR) na superfície dos hepatócitos para promover a degradação destes receptores dentro do fígado. O LDLR é o receptor primário responsável pela remoção das LDLs circulantes. Portanto, a redução do número de LDLR pela PCSK9 resulta em um maior nível sanguíneo de LDL-C. Ao inibir a ligação da PCSK9 ao LDLR, o alirocumabe aumenta o número de receptores disponíveis para remover as LDLs circulantes, diminuindo desta forma os níveis de LDL-C.
O LDLR também se liga aos remanescentes das lipoproteínas ricas em triglicérides e às lipoproteínas de densidade intermediária (IDL). Portanto, o tratamento com alirocumabe pode produzir reduções nessas lipoproteínas, como evidenciado pelas reduções de Apo B, colesterol não HDL e TGs. O alirocumabe também resulta em reduções na Lp(a), que é uma forma de LDL que está ligado a apolipoproteína (a). Entretanto, o LDLR demonstrou possuir uma baixa afinidade pela Lp(a), portanto o mecanismo exato pelo qual o alirocumabe diminui a Lp(a) não é ainda totalmente compreendido.
Em estudos genéticos em humanos foram identificadas variantes de PCSK9 com mutações tanto com ganho quanto com perda de função. Indivíduos com mutações em um único alelo da PCSK9 promovendo perda de função possuem níveis mais baixos de LDL-C, o que se correlacionou com uma incidência mais baixa de doença cardíaca coronariana. Foram relatados poucos indivíduos que possuíam mutações nos dois alelos da PCSK9 promovendo perda de função e que possuíam níveis profundamente baixos de LDL-C com níveis de HDL-C e TG na faixa normal. Por outro lado, mutações de ganho de função no gene PCSK9 foram identificadas em pacientes com níveis aumentados de LDL-C e diagnóstico clínico de hipercolesterolemia familiar.
Análises observacionais demonstraram que o não tratamento de níveis elevados de LDL-C em pacientes com mutações com ganho de função no gene PCSK9 estão em uma faixa similar àqueles observados em pacientes com as mutações mais tradicionais que causam HFHe (tais como no gene do LDLR) demonstrando um papel central da PCSK9 no metabolismo e nos níveis do LDL-C. Em um estudo multicêntrico, duplo-cego, controlado com placebo, de 14 semanas, 13 pacientes com HFHe com ganho de função devido à mutações no gene da PCSK9 foram randomizados para receber alirocumabe 150 mg uma vez a cada duas semanas ou placebo. O LDL-C médio basal era 151,5 mg/dL. Na semana 2, a redução média em relação ao LDL-C basal foi 62,5% nos pacientes tratados com alirocumabe em comparação a 8,8% nos pacientes recebendo placebo. Na semana 8, a redução média de LDL-C a partir do basal em todos os pacientes tratados com alirocumabe foi de 72,4%.
Propriedades farmacodinâmicas
O alirocumabe é um anticorpo monoclonal totalmente humano que inibe a atividade da PCSK9 tanto nos testes in vitro como em modelos in vivo. Um conjunto de estudos em animais e seres humanos demonstrou o papel central que níveis elevados de LDL-C desempenham na iniciação e progressão da aterosclerose.
Outras lipoproteínas que contem Apo B-100, especificamente os remanescentes de lipoproteínas ricas em triglicérides (derivados das VLDLs e IDLs) e a Lp(a), promovem a aterosclerose. Entretanto, até o momento não foi determinado o efeito independente da redução dessas lipoproteínas na morbidade e mortalidade cardiovascular.
Nos ensaios in vitro, o alirocumabe não induziu a atividade de função efetora mediada por Fc (toxicidade mediada por células dependentes de anticorpos e citotoxicidade dependente de complemento) tanto na presença como na ausência de PCSK9, e não foram observados complexos imunes solúveis capazes de ligar-se a proteínas de complemento para o alirocumabe quando ligado a PCSK9.
Propriedades farmacocinéticas
Absorção
Após administração subcutânea (SC) de 50 mg a 300 mg de alirocumabe, o tempo médio para atingir concentração sérica máxima (tmax) foi entre 3 e 7 dias.
A farmacocinética do alirocumabe após administração SC única de 75 mg no abdômen, parte superior do braço ou na coxa foi similar.
A biodisponibilidade absoluta do alirocumabe após administração SC foi aproximadamente 85% como determinado pela análise farmacocinética da população.
Observou-se um aumento um pouco maior do que o proporcional à dosagem, com um aumento de 2,1 a 2,7 vezes nas concentrações totais de alirocumabe com o aumento de 2 vezes na dosagem, de 75 mg para 150 mg uma vez a cada duas semanas.
O estado de equilíbrio (steady state) foi alcançado após 2 a 3 doses com uma taxa de acúmulo até um máximo de cerca de 2 vezes.
Distribuição
Após administração IV, o volume de distribuição foi de cerca de 0,04 a 0,05 L/kg, indicando que o alirocumabe é distribuído principalmente no sistema circulatório.
Metabolismo
Estudos de metabolismo específicos não foram conduzidos, pois o alirocumabe é uma proteína.
Espera-se que o alirocumabe seja degradado em pequenos peptídeos e aminoácidos individuais.
Eliminação
Foram observadas duas fases de eliminação de alirocumabe. Em baixas concentrações, a eliminação é predominantemente através de ligação saturável ao alvo (PCSK9), enquanto em concentrações mais altas a eliminação do alirocumabe é em grande parte através de uma via proteolítica não saturável.
Baseado em uma análise farmacocinética populacional, a meia-vida do alirocumabe no estado de equilíbrio foi de 17 a 20 dias em pacientes recebendo alirocumabe como monoterapia em doses subcutâneas de 75 mg uma vez a cada duas semanas ou 150 mg uma vez a cada duas semanas. Quando coadministrado com uma estatina, a meia-vida do alirocumabe foi de 12 dias.
Populações especiais
Gênero
Baseado na análise farmacocinética da população, o sexo não teve impacto na farmacocinética de alirocumabe.
Pacientes idosos
Baseado na análise farmacocinética da população, a idade esteve associada a uma pequena diferença na exposição ao alirocumabe no estado de equilíbrio, sem impacto na eficácia ou segurança.
Peso corpóreo
Baseado na análise farmacocinética da população, o peso corporal teve pequeno impacto na exposição ao alirocumabe, sem impacto na eficácia ou segurança.
Pacientes pediátricos
Os efeitos farmacocinéticos da administração de alirocumabe em pacientes pediátricos não foram estudados.
Insuficiência hepática
Em um estudo fase 1, após administração de uma dose única SC de 75 mg, o perfil farmacocinético do alirocumabe em indivíduos com insuficiência hepática leve e moderada foi similar à comparada a indivíduos com função hepática normal. Não estão disponíveis dados em pacientes com insuficiência hepática severa.
Insuficiência renal
Uma vez que não há evidência de excreção renal dos anticorpos monoclonais, não se espera que a função renal tenha impacto na farmacocinética de alirocumabe. Análise de farmacocinética da população demonstrou que insuficiência renal leve e moderada não teve um impacto significativo na farmacocinética do alirocumabe. Não há dados disponíveis em relação a pacientes com insuficiência renal severa.
Raça
Baseado na análise farmacocinética da população, a raça não teve impacto na farmacocinética de alirocumabe.
Após administração SC de dose única de 100 mg a 300 mg de alirocumabe, não houve diferença significativa na exposição entre indivíduos saudáveis japoneses e caucasianos.
Relação farmacocinética – farmacodinâmica
O efeito farmacodinâmico de alirocumabe em diminuir LDL-C é indireto e mediado pela inibição da PCSK9. A redução do LDL-C dependente da concentração de PCSK9 é observada até que a saturação alvo seja atingida. Após a saturação da ligação à PCSK9, o aumento adicional nas concentrações de alirocumabe não resulta em reduções subsequentes de LDL-C, entretanto, é observada uma duração estendida do efeito de redução de LDL-C.
Dados de segurança não clínicos
Farmacologia animal
O alirocumabe se liga com alta afinidade à PCSK9 em várias espécies e bloqueia sua interação com o LDLR in vitro e in vivo. A administração de alirocumabe leva a redução do LDL-C de maneira dosedependente em diferentes modelos animais, incluindo camundongos, ratos, hamsters e macacos.
Em um estudo de aterosclerose em camundongos APOE*3Leiden.CETP, uma diminuição dose dependente no colesterol e nos triglicérides séricos totais, e um aumento nos níveis de LDLR hepáticos foram observados com dosagens repetidas de alirocumabe (3 ou 10 mg/kg/semana SC), comparado com camundongos controle. Além disso, o alirocumabe diminuiu de maneira dose-dependentemente o tamanho das lesões ateroscleróticas, e melhorou marcadores de estabilidade das placas.
Toxicidade aguda
Não foram realizados estudos toxicológicos de dose única.
Toxicidade crônica
Estudos toxicológicos de dose repetida foram conduzidos em ratos Sprague-Dawley e macacos cinomolgos. Nos estudos toxicológicos de 26 semanas, os NOAELs foram as doses máximas administradas, 50 mg/kg/semana SC (subcutâneo) em ratos e 75 mg/kg/semana SC nos macacos, correspondendo a aproximadamente 11 vezes e 103 vezes, respectivamente, a exposição humana na dosagem de 150 mg uma vez a cada duas semanas baseado na área sob a curva (ASC).
Carcinogenicidade
Estudos de carcinogenicidade não foram conduzidos com alirocumabe.
Mutagênese
Estudos de mutagênese não foram conduzidos com alirocumabe.
Genotoxicidade
Estudos de genotoxicidade não foram conduzidos com alirocumabe.
Teratogenicidade/desenvolvimento embriofetal
Em estudos toxicológicos de desenvolvimento em ratos e macacos, quando fêmeas grávidas foram expostas ao alirocumabe, concentrações quantificáveis de alirocumabe no soro foram observadas nos fetos de ratos e macacos, indicando que o alirocumabe, como outros anticorpos IgG, atravessam a barreira placentária.
Não houve efeitos sobre o crescimento ou desenvolvimento fetal no estudo de desenvolvimento fetoembrionário em ratos realizados com doses até 75 mg/kg/dose administrados nos dias de gestação 6 e 12. Em um estudo toxicológico de 5 semanas, a administração semanal de 75 mg/kg a ratas não grávidas resultou em exposições sistêmicas que eram aproximadamente 5,3 vezes a exposição humana na dosagem de 150 mg uma vez a cada duas semanas, baseado na ASC. Em macacos, uma resposta imunológica humoral secundária mais fraca ao desafio do antígeno foi observada em filhotes de macacos quando o alirocumabe foi administrado durante a organogênese até o parto em exposições de dose 13 vezes a exposição máxima recomendada para humanos de 150 mg uma vez a cada duas semanas. Nenhum efeito adicional durante a gestação ou no desenvolvimento neonatal/recém-nascido foi observado com exposições de dose 81 vezes a dose máxima recomendada para humanos de 150 mg uma vez a cada duas semanas.
Diminuição da fertilidade
Não houve eventos adversos em marcadores de fertilidade (p.ex. ciclicidade estral, volume testicular, volume de ejaculação, motilidade dos espermatozoides, ou contagem total de espermatozoides por ejaculação) em um estudo de toxicidade crônica de 26 semanas em macacos sexualmente maduros. Além disso, não houve alterações anatômicas relacionadas ao alirocumabe ou achados histopatológicos em tecidos reprodutivos em qualquer rato ou macaco avaliado em estudo toxicológico.