 Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)
Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)Qual a ação da substância do Iclusig?
Resultados de Eficácia
A eficácia e segurança do Cloridrato de Ponatinibe em pacientes com LMC e LLA Ph+ que foram resistentes ou intolerantes ao tratamento anterior com inibidor da tirosina quinase (ITQ) foram avaliadas em um estudo clínico multicêntrico de um único braço, aberto, internacional. Todos os pacientes receberam 45 mg de Cloridrato de Ponatinibe uma vez ao dia com a possibilidade de diminuições da dose e interrupções da dose seguidas pela retomada e aumento da dose. Os pacientes foram atribuídos a um de seis grupos com base na fase da doença (LMC-FC; LMC-FA; ou LMC-FB/LLA Ph+), resistentes ou intolerantes (R/I) ao dasatinibe ou nilotinibe, e presença da mutação T315I. O estudo está em andamento.
A resistência em LMC - FC foi definida como falha em se obter quer resposta hematológica completa (em 3 meses), resposta citogenética menor (em 6 meses), ou resposta citogenética maior (em 12 meses) durante o tratamento com dasatinibe ou nilotinibe. Pacientes com LMC-FC que experimentaram uma perda de resposta ou desenvolvimento de uma mutação no domínio da quinase na ausência de uma resposta citogenética completa ou progressão para LMC-FA ou LMC-FB a qualquer altura do tratamento com dasatinibe ou nilotinibe também foram considerados resistentes. A resistência em LMC-FA e LMC- FA/LLA Ph+ foi definida como uma falha em obter quer uma resposta hematológica maior (LMC-FA em 3 meses, LMC-FB/LLA Ph+ em 1 mês), perda de resposta hematológica maior (a qualquer altura), ou desenvolvimento de mutação de domínio de quinase na ausência de uma resposta hematológica maior durante o tratamento com dasatinibe ou nilotinibe.
A intolerância foi definida como a descontinuação do dasatinibe ou nilotinibe devido a toxicidades apesar da gestão ótima na ausência de uma resposta citogenética completa para pacientes com LMCFC ou resposta hematológica maior para pacientes com LMC-FA, LMC-FB, ou LLA Ph+.
O desfecho primário de eficácia em LMC-FC foi a resposta citogenética maior (RCyM), que incluiu respostas citogenéticas completas e parciais (RCyC e PCyR). Os desfechos secundários de eficácia em LMC-FC foram a resposta hematológica completa (RHC) e a resposta molecular maior (RMM).
O desfecho primário em LMC-FA e LMC-FB/LLA Ph+ foi a resposta hematológica maior (RHM), definida quer como uma resposta hematológica completa (RHC) ou sem evidência de leucemia (SEL). Os parâmetros de avaliação final secundários em LMC-FA e LMC-FB/LLA Ph+ foram RCyM e RMM.
Para todos os pacientes, desfechos secundários de eficácia adicionais incluíram:
RCyM confirmada, tempo até resposta, duração da resposta, sobrevida livre de progressão (SLP) e sobrevida global (SG). Também foram realizadas análises de acompanhamento (“post -hoc”) para avaliar a relação de resultados de resposta citogenética (RCyM) e molecular (RMM) de curto prazo com os resultados de SLP e SG de longo prazo, manutenção da resposta (RCyM e RMM) após reduções da dose, e SLP e SG por estado de Evento de Oclusão Arterial.
No estudo, foram recrutados 449 pacientes nos quais 444 foram elegíveis para análise:
- 267 pacientes com LMC-FC (Grupo R/I: n=203, Grupo T315I: n=64), 83 pacientes com LMC-FA (Grupo R/I: n=65, Grupo T315I: n=18), 62 pacientes com LMC-FB (Grupo R/I: n=38, Grupo T315I: n=24) e 32 pacientes com LLA Ph+ (Grupo R/I: n=10, Grupo T315I: n=22). Uma RCyM anterior ou melhor (RCyM, RMM ou RMC) ao dasatinibe ou nilotinibe apenas foi obtida em 26% dos pacientes com LMC-FC e uma anterior RHM ou melhor (RHM, RCyM, RMM ou RMC) foi obtida apenas em 21%, e 24% dos pacientes com LMC-FA e LMC-FB/LLA Ph+, respectivamente. As características demográficas do início estão descritas na tabela 1 abaixo.
Tabela 1: Características demográficas e da doença:
| Características dos pacientes no início | Eficácia População Total N = 449 |
| Idade | |
| Média, anos (intervalo | 59 (18 a 94) |
| Sexo, n (%) | |
| Masculino | 238 (53%) |
| Raça, n (%) | |
| Asiática | 59 (13%) |
| Negra ou afro-americana | 25 (6%) |
| Branca | 352 (78%) |
| Outras | 13 (3%) |
| Índice de desempenho ECOG, n (%) | |
| ECOG = 0 or 1 | 414 (92%) |
| Histórico de doença | |
| Tempo médio do diagnóstico a primeira dose, anos (intervalo) | 6,09 (0,33 - 28,47) |
| Resistência a terapia anterior com ITQa *, n (%) | 374 (88%) |
| Terapia prévia com ITQ – número de regimes, n (%) | 244 (55%) |
| Terapia prévia com ITQ – número de ITOs prévios aprovados, n (%) | --- |
| 1 | 32 (7%) |
| 2 | 155 (35%) |
| ≥3 | 262 (58%) |
| Mutação BCR-ABL detectada no início, n (%)b | --- |
| Nenhum | 198 (44%) |
| 1 | 192 (43%) |
| ≥2 | 54 (12%) |
a* De 427 pacientes que reportaram tratamento anterior IQT com dasatinibe ou nilotinibe.
b Dos pacientes com uma ou mais mutações do domínio BCR-ABL quinase detectadas na inclusão, foram detectadas 37 mutações únicas.
No geral, 55% dos pacientes tinham uma ou mais mutações no domínio da quinase BCR-ABL no início sendo as mais frequentes:
- T315I (29%), F317L (8%), E255K (4%) e E359V (4%). Em 67% dos pacientes com LMC-FC no grupo R/I não foram detectadas mutações no início do estudo.
Os resultados de eficácia estão resumidos nas tabelas 2, 3 e 4.
Tabela 2: Eficácia de Cloridrato de Ponatinibe em pacientes com LMC em Fase Crônica resistentes ou intolerantes:
| --- | Total (N = 267) | Resistente ou Intolerante | |
| Resposta Citogenética | Coorte R/I (N=203) | Coorte T315I (N= 64) | |
| Maiora (RCyR)% (IC de 95%) | 55% (50-62) | 51% (44-58) | 70% (58-81) |
| Completa (CCyR)% (IC de 95%) | 46% (40-52) | 40% (33-47) | 66% (53-77) |
| Resposta Molecular Maiorb % (IC de 95%) | 40% (35-47) | 35% (28-42) | 58% (45-70) |
aDesfecho primário para as coortes LMC-FC foi RCyM, que combina ambas respostas citogenéticas tanto completas (sem células Ph+ detectáveis) como parciais (1% a 35% de células Ph+).
b Medido em sangue periférico. Definido como taxa ≤0,1% de transcrições BCR-ABL a ABL na Escala Internacional (IS) (ie, ≤0,1% BCR-ABLIS; os pacientes devem ter a transcrição b2a2/b3a2 (p210)), medido no sangue periférico por reação em cadeia de polimerase com transcriptase reversa quantitativa (qRT PCR).
Retirada da base de dados de 06 de fevereiro de 2017.
Os pacientes com LMC-FC que receberam menos ITQs anteriormente obtiveram respostas citogenéticas, hematológicas e moleculares maiores. Dos pacientes com LMC-FC previamente tratados com um, dois, três ou quatro ITQs anteriores, 75% (12/16), 68% (66/97), 44% (63/142) e 58% (7/12) obtiveram uma RCyM durante o tratamento com Cloridrato de Ponatinibe, respectivamente.
Dos pacientes com LMC-FC sem mutação detectada no início, 49% (66/136) obtiveram uma RCyM.
Para cada mutação BCR-ABL detectada em mais do que um paciente com LMC-FC no início, obteve-se uma RCyM no seguimento ao tratamento com Cloridrato de Ponatinibe.
Em pacientes com LMC-FC que obtiveram uma RCyM, o tempo médio para RCyM foi de 2,8 meses (intervalo: 1,6 a 11,3 meses) e em pacientes que alcançaram uma RMM, o tempo médio para RMM foi de 5,5 meses (intervalo: 1,8 a 55,5 meses). Na altura da comunicação atualizada, com acompanhamento mínimo de 64 meses para todos os pacientes no estudo, as durações médias de RCyM e de RMM ainda não haviam sido obtidas. Com base nas estimativas de Kaplan-Meier, 82% (95% IC: [74%–88%]) dos pacientes com LMC-FC (mediana de duração do tratamento: 32,2 meses) que obtiveram uma RCyM foram projetados para manter essa resposta aos 48 meses e 61% (95% IC: [51%-70%]) dos pacientes com LMC-FC que obtiveram uma RMM estão projetados para manter essa resposta aos 36 meses. A probabilidade de todos os pacientes com LMC-FC manterem a RCyM e RMM não mudou após a análise ter sido aumentada para 5 anos.
Com um acompanhamento mínimo de 64 meses, 3,4% (9/267) dos pacientes com LMC-FC apresentaram a evolução da doença para LMC-FA ou LMC-FB.
Para o total de pacientes com LMC-FC (N=267), bem como para os pacientes com LMC-FC R/I do Grupo A R/I (N = 203) e do Grupo B T315I (N=64), a mediana da SG ainda não foi atingida. Para o grupo de pacientes LMC-FC, a probabilidade de sobrevivência a 2, 3, 4 e 5 anos é estimada em 86,0%, 81,2%, 76,9% e 73,3%, respectivamente, como demonstrado na Figura 1.
Figura 1- Estimativas de Kaplan-Meier referentes à sobrevida global na população com LMCFC (População Tratada):
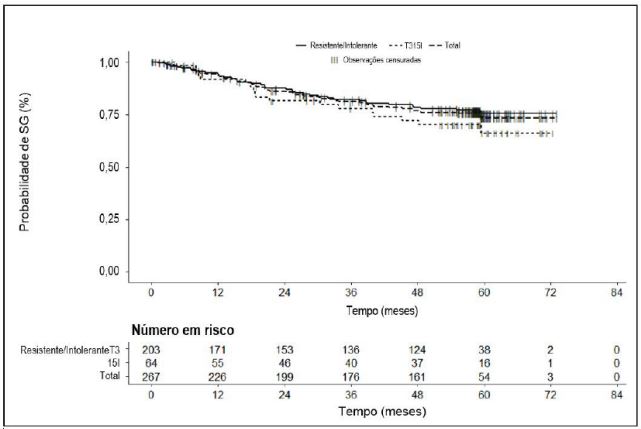
Os pacientes com LMC-FC que obtiveram uma resposta de RCyM ou RMM no primeiro ano de tratamento tiveram uma melhoria estatisticamente significativa da sobrevida livre de progressão (SLP) e sobrevida global (SG) em comparação com os pacientes que não cumpriram estes marcos de tratamento. Uma RCyM no período de 3 meses foi correlacionada fortemente e de forma estatisticamente significativa com uma SLP e SG (p<0,0001 e p=0,0006, respectivamente). A significância estatística foi obtida na correlação de SLP e SG com uma RCyM no período de 12 meses (p= < 0,0001 e p=0,0012, respectivamente).
Tabela 3: Eficácia de Cloridrato de Ponatinibe em pacientes com LMC fase avançada resistente ou intolerante:
| Taxa de resposta hematológica | LMC de fase acelerada | LMC de fase blástica | ||||
| Total (N=83) | Resistente ou Intolerante | Geral (N=62) | Resistente ou Intolerante | |||
| Coorte R/I (N=65) | Coorte T315I (N=18) | Coorte R/I (N=38) | Coorte T315I (N=24) | |||
| Maiora (RHM) % (IC de 95%) | 57% (45-68) | 57% (44-69) | 56% (31-79) | 31% (20–44) | 32% (18–49) | 29% (13–51) |
| Completab (RHC) % (IC de 95%) | 51% (39-62) | 49% (37-62) | 56% (31-79) | 21% (12-33) | 24% (11-40) | 17% (5-37) |
| Resposta citogenética maiorc % (IC de 95%) | 39% (28-50) | 34% (23-47) | 56% (31-79) | 23% (13-35) | 18% (8-34) | 29% (13-51) |
a Desfecho primário para as coortes LMC-FA e LMC-FB/LLA Ph+ foi a RHM que combina as respostas hematológicas completas e ausência de sinais de leucemia.
b RHC: WBC ≤ ULN institucional, ANC ≥1000/mm3, plaquetas ≥100.000/mm3, ausência de blastos ou promielócitos no sangue periférico, blastos na medula ≤5%, <5% de mielócitos mais metamielócitos no sangue periférico, basófilos <5% no sangue periférico, sem envolvimento extramedular (incluindo sem hepatomegalia ou esplenomegalia).
c A RCyM combina tanto respostas citogenéticas completas (sem células Ph+ detectáveis) como parciais (1% a 35% de células Ph+).
Retirada da base de dados em 6 de fevereiro de 2017.
Tabela 4: Eficácia do Cloridrato de Ponatinibe em pacientes com LLA Ph+ resistentes ou intolerantes:
| Total (N = 32) | Resistente ou Intolerante | ||
| Coorte R/I (N=10) | Coorte T315I (N= 22) | ||
| Taxa Resposta Hematológica | --- | --- | --- |
| Maiora (RHM)% (IC de 95%) | 41% (24-59) | 50% (19-81) | 36% (17-59) |
| Completab (CCyR)% (IC de 95%) | 34% (19-53) | 40% (12-74) | 32% |
| Resposta Molecular Maiorc % (IC de 95%) | 47% (29-65) | 60% (26-88) | 41% (21-64) |
a Desfecho primário para as coortes LMC-FA e LMC-FB/LLA Ph+ foi a RHM que combina as respostas hematológicas completas e ausência de sinais de leucemia.
b RHC: WBC ≤ ULN institucional, ANC ≥1000/mm3 , plaquetas ≥100.000/mm3 , ausência de blastos ou promielócitos no sangue periférico, blastos na medula ≤5%, <5% de mielócitos mais metamielócitos no sangue periférico, basófilos <5% no sangue periférico, sem envolvimento extramedular (incluindo sem hepatomegalia ou esplenomegalia).
c A RCyM combina tanto respostas citogenéticas completas (sem células Ph+ detectáveis) como parciais (1% a 35% de células Ph+).
Retirada da base de dados em 6 de fevereiro de 2017.
O tempo médio até RHM em pacientes com LMC-FA, LMC-FB e LLA Ph+ foi de 0,7 meses (intervalo: 0,4 a 5,8 meses), 1,0 meses (intervalo: 0,4 a 3,7 meses) e 0,7 meses (intervalo: 0,4 a 5,5 meses), respectivamente. Até o momento da comunicação atualizada, com acompanhamento mínimo de 48 meses para todos os pacientes no estudo, a duração média da RHM para pacientes com LMC-FA (duração média do tratamento: 19,4 meses), LMC-FB (mediana de duração do tratamento: 2,9 meses) e LLA Ph+ (duração média do tratamento: 2,7 meses) foi estimada em 12,9 meses (intervalo: 1,2 a 52+ meses), 6,0 meses (intervalo: 1,8 a 47,4+ meses) e 3,2 meses (intervalo: 1,8 a 12,8+meses), respectivamente.
Para todos os pacientes em estudo de fase 2, a relação intensidade-segurança da dose indicou a existência de aumentos significativos nos eventos adversos de grau ≥ 3 (insuficiência cardíaca, trombose arterial, hipertensão, trombocitopenia, pancreatite, neutropenia, erupção cutânea, aumento de ALT, aumento de AST, aumento da lipase, mielosupressão, artralgia) no intervalo posológico de 15 a 45 mg uma vez ao dia.
A análise da relação intensidade-segurança da dose no estudo de fase 2 concluiu que após o ajuste para as covariáveis, a intensidade geral da dose é significativamente associada a um risco elevado de oclusão vascular, com um índice de probabilidades de aproximadamente 1,6 para cada aumento de 15 mg. Adicionalmente, os resultados de análises de regressão logística de dados de pacientes incluídos no estudo de fase 1 sugerem uma relação entre a exposição sistêmica (AUC) e a ocorrência de eventos trombóticos arteriais. Por conseguinte, prevê-se que uma redução da dose reduza o risco de eventos oclusivos vasculares. No entanto, a análise sugeriu que pode haver um efeito de “transferência” de doses mais elevadas, de tal modo que pode demorar vários meses até uma redução da dose se manifestar em redução do risco. As outras covariáveis que demostram uma associação estatisticamente significativa com a ocorrência de eventos oclusivos vasculares, nesta análise, são a história clínica de isquemia e a idade.
Redução da dose em pacientes com LMC-FC
No estudo de fase 2, foram recomendadas reduções de dose no seguimento de eventos adversos. Adicionalmente, em outubro de 2013, foram introduzidas neste ensaio novas recomendações para a redução prospectiva da dose em todos os pacientes com LMC-FC na ausência de eventos adversos, com o objetivo de reduzir o risco de eventos oclusivos vasculares.
Com acompanhamento mínimo de 48 meses e aproximadamente 2 anos após a recomendação para a redução prospectiva da dose, havia 110 pacientes com LMC-FC em curso. Foi comunicado que a maioria destes pacientes em curso (82/110, 75%) receberam 15 mg na última dose, enquanto 24/110 pacientes (22%) receberam 30 mg e 4/110 (4%) receberam 45 mg. No momento do início do fechamento do estudo (acompanhamento mínimo de 64 meses e mais de 3 anos após a recomendação para redução prospectiva da dose), 99 pacientes com LMC-FC estavam em andamento e 77 (78%) desses pacientes receberam 15 mg como última dose no estudo.
Segurança
No estudo de fase 2, 86 pacientes com LMC-FC alcançaram uma RCyM com uma dose de 45 mg, 45 pacientes com LMC-FC alcançaram uma RCyM após uma redução da dose para 30 mg, a maioria devido a eventos adversos.
Eventos oclusivos vasculares ocorreram em 44 destes 131 pacientes. A maioria destes eventos ocorreu com a dose em que o paciente alcançou uma RCyM; ocorreram menos eventos após a redução da dose.
Tabela 5: Primeiros eventos adversos vasculares oclusivos em pacientes com LMC-FC que alcançaram uma RCyM com a dose de 45 mg ou 30 mg (extração de dados a 7 de abril de 2014):
| --- | Dose mais recente desde o início do primeiro evento vascular oclusivo | ||
| 45 mg | 30 mg | 15 mg | |
| Alcançaram RCyM com 45 mg (N=86) | 19 | 6 | 0 |
| Alcançaram RCyM com 30 mg (N=45) | 1 | 13 | 5 |
O tempo médio até ao início dos primeiros eventos cardiovasculares, cerebrovasculares e de oclusão arterial vascular periférica foi de 351,611 e 605 dias, respectivamente. Quando ajustado em relação à exposição, a incidência dos primeiros eventos oclusivos arteriais foi superior nos primeiros dois anos de acompanhamento e diminuiu com a redução da intensidade da dose diária (após recomendação para a redução prospectiva da dose). Outros fatores diferentes da dose podem também contribuir para este risco de oclusão arterial.
Eficácia
Estão disponíveis dados preliminares do ensaio de fase 2 sobre a manutenção de resposta (RCyM e RMM) em todos os pacientes com LMC-FC submetidos a redução da dose por qualquer motivo. O quadro 6 mostra estes dados de pacientes que alcançaram RCyM e RMM com a dose de 45 mg; estão disponíveis dados similares para pacientes que alcançaram RCyM e RMM com a dose de 30 mg. A maioria dos pacientes submetidos à redução da dose manteve a resposta (RCyM e RMM) ao longo da duração do acompanhamento atualmente disponível. Uma percentagem de pacientes não foi submetida a qualquer redução da dose, com base na respectiva avaliação benefício-risco.
Tabela 6: Manutenção de resposta em pacientes com LMC-FC que alcançaram RCyM ou RMM com a dose de 45 mg (extração de dados em 6 de fevereiro de 2017):
| Alcançaram RCyM com 45 mg (N=86) | Alcançaram RMM com 45 mg (N=63) | |||
| Número de pacientes | Mantiveram RCyM | Número de pacientes | Mantiveram RMM | |
| Sem redução da dose | 19 | 13 (68%) | 18 | 11 (61%) |
| Redução da dose para 30 mg apenas | 15 | 13(87%) | 5 | 3 (60%) |
| Redução ≥ 3 meses para 30 mg | 12 | 10 (83%) | 3 | 2 (67%) |
| Redução ≥ 6 meses para 30 mg | 11 | 9 (82%) | 3 | 2 (67%) |
| Redução ≥ 12 meses para 30 mg | 8 | 7 (88%) | 3 | 2 (67%) |
| Redução ≥ 18 meses para 30 mg | 7 | 6 (100%) | 2 | 2 (100%) |
| Redução ≥ 24 meses para 30 mg | 6 | 1 (100%) | --- | --- |
| Redução ≥ 36 meses para 30 mg | 1 | 1 (100%) | --- | --- |
| Qualquer redução da dose para 15 mg | 52 | 51 (98%) | 40 | 36 (90%) |
| Redução ≥ 3 meses para 15 mg | 49 | 49 (100%) | 39 | 36 (92%) |
| Redução ≥ 6 meses para 15 mg | 47 | 47 (100%) | 37 | 35 (95%) |
| Redução ≥ 12 meses para 15 mg | 44 | 44 (100%) | 34 | 33 (97%) |
| Redução ≥ 18 meses para 15 mg | 38 | 38 (100%) | 29 | 29 (100%) |
| Redução ≥ 24 meses para 15 mg | 32 | 32 (100%) | 23 | 23 (100%) |
| Redução ≥ 36 meses para 15 mg | 8 | 8 (100%) | 4 | 4 (100%) |
A atividade anti-leucêmica do Cloridrato de Ponatinibe foi igualmente avaliada num estudo de escalonamento da dose de fase 1 que incluiu 65 pacientes com LMC e LLA Ph+; o estudo está em curso. Dos 43 pacientes com LMC-FC, 31 pacientes com LMC-FC alcançaram uma RCyM com uma duração média de acompanhamento de 55,5 meses (intervalo: 1,7 a 91,4 meses). dano momento da comunicação, 25 pacientes com LMC-FC estavam em RCyM (a duração média da RCyM ainda não tinha sido obtida).
Eletrofisiologia cardíaca
O potencial de prolongamento do intervalo QT de Cloridrato de Ponatinibe foi avaliado em 39 pacientes com leucemia que receberam 30 mg, 45 mg, ou 60 mg de Cloridrato de Ponatinibe uma vez ao dia. Foram coletados ECGs triplicatas, em série, no início e em estado estável para avaliar o efeito do Cloridrato de Ponatinibe nos intervalos QT. Não foram detectadas alterações clinicamente significativas no intervalo QTc médio (i.e., > 20 ms) em relação ao início do estudo. Além disso, os modelos farmacocinéticos-farmacodinâmicos não revelam qualquer relação exposição-efeito, com uma alteração média de QTcF estimada de –6,4 ms (intervalo de confiança superior –0,9 ms) na Cmax para o grupo dos 60 mg.
Características Farmacológicas
Farmacodinâmica
O Cloridrato de Ponatinibe é um potente inibidor pan BCR-ABL com elementos estruturais, incluindo uma ligação tripla carbono-carbono, que permitem a ligação de elevada afinidade ao BCR-ABL nativo e formas mutantes de ABL quinase. O Cloridrato de Ponatinibe inibe a atividade tirosina quinase do ABL e do mutante T315I de ABL com valores IC50 de 0,4 e 2,0 nM, respectivamente. Em estudos celulares, o Cloridrato de Ponatinibe superou a resistência mediada por mutações no domínio da quinase BCR-ABL do imatinibe, dasatinibe e nilotinibe.
Em estudos pré-clínicos de mutagênese, 40 nM foi determinada como a concentração do Cloridrato de Ponatinibe suficiente para inibir a viabilidade das células que expressam todos os mutantes BCR-ABL testados em > 50% (incluindo T315I) e suprimir o surgimento de clones mutantes. Num estudo celular de mutagênese acelerada, não foi detectada nenhuma mutação em BCR-ABL que pudesse conferir resistência a 40 nM de Cloridrato de Ponatinibe.
O Cloridrato de Ponatinibe favoreceu a redução do tumor e prolongou a sobrevida em camundongos com tumores que expressam BCR-ABL nativo ou T315I mutante.
Em doses de 30 mg ou superiores, as concentrações no plasma de Cloridrato de Ponatinibe no estado de equilíbrio estável excedeu tipicamente 21 ng/mL (40 nM).
Em doses de 15 mg ou superiores, 32 de 34 pacientes (94%) demonstraram uma redução ≥ 50% na fosforilação da CRK-like (CRKL), um bio- marcador da inibição BCR-ABL nas células mononucleares do sangue periférico.
O Cloridrato de Ponatinibe inibe a atividade de outras quinases clinicamente relevantes com valores IC50 abaixo de 20 nM e demonstrou atividade celular contra a RET, FLT3, e KIT e membros das famílias FGFR, PDGFR e VEGFR de quinases.
Farmacocinética
Absorção
As concentrações máximas de Cloridrato de Ponatinibe são observadas aproximadamente 4 horas após a administração oral. Dentro do intervalo de doses clinicamente relevantes avaliadas em pacientes (15 mg a 60 mg), o Cloridrato de Ponatinibe exibiu aumentos proporcionais da dose tanto em Cmax como AUC. A média geométrica (CV%) das exposições Cmax e AUC(0-τ) obtidas para 45 mg de Cloridrato de Ponatinibe diários em estado de equilíbrio estacionário foram de 77 ng/mL (50%) e 1296 ng•hr/mL (48%), respectivamente. A exposição a uma dieta rica em gorduras ou uma dieta pobre em gorduras, as exposições plasmáticas do Cloridrato de Ponatinibe (Cmáx e AUC) não foram diferentes das condições em jejum. Cloridrato de Ponatinibe pode ser administrado com ou sem alimentos. A administração concomitante de Cloridrato de Ponatinibe com um inibidor potente da secreção ácida gástrica resultou numa pequena redução da Cmax de Cloridrato de Ponatinibe sem redução da AUC0-∞.
Distribuição
O Cloridrato de Ponatinibe tem uma elevada ligação (> 99%) as proteínas plasmáticas in vitro. A razão sangue/plasma do Cloridrato de Ponatinibe é de 0,96. O Cloridrato de Ponatinibe não é deslocado pela administração concomitante de ibuprofeno, nifedipino, propranolol, ácido salicílico ou varfarina. Com doses diárias de 45 mg, a média geométrica (CV%) do volume de distribuição em estado de equilíbrio estacionário aparente é de 1101 L (94%) sugerindo que o Cloridrato de Ponatinibe é extensivamente distribuído no espaço extravascular. Estudos in vitro sugeriram que o Cloridrato de Ponatinibe ou não é um substrato ou é um substrato fraco para a P-gp e para a proteína de resistência do câncer da mama BCRP. O Cloridrato de Ponatinibe não é um substrato para os polipeptídios humanos transportadores de anions orgânicos, OATP1B1, OATP1B3, e ao transportador de cátions orgânicos, OCT-1.
Biotransformação
O Cloridrato de Ponatinibe é metabolizado num ácido carboxílico inativo por esterases e/ou amidases e é metabolizado pelo CYP3A4 em um metabólito de N-desmetil que é 4 vezes menos ativo do que o Cloridrato de Ponatinibe. O ácido carboxílico e o metabolito N-desmetil compreendem 58% e 2% dos níveis de Cloridrato de Ponatinibe em circulação, respectivamente.
Nas concentrações séricas terapêuticas, o Cloridrato de Ponatinibe não inibiu OATP1B1 ou OATP1B3, OCT1 ou OCT2, os transportadores de anions orgânicos OAT1 ou OAT3, ou a bomba de exportação de sais biliares (BSEP) in vitro. Por esse motivo, é pouco provável que ocorram interações medicamentosas clínicas como resultado da inibição dos substratos mediada por Cloridrato de Ponatinibe para estes transportadores. Os estudos in vitro indicam que há pouca probabilidade de ocorrerem interações medicamentosas clínicas em resultado da inibição mediada pelo Cloridrato de Ponatinibe do metabolismo dos substratos relativos a CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP3A ou CYP2D6.
Um estudo in vitro em hepatócitos humanos indicou que é igualmente pouco provável ocorrerem interações medicamentosas clínicas em resultado de indução mediada pelo Cloridrato de Ponatinibe do metabolismo dos substratos relativos a CYP1A2, CYP2B6 ou CYP3A.
Eliminação
No seguimento de doses únicas e múltiplas de 45 mg de Cloridrato de Ponatinibe, a meia vida de eliminação terminal do Cloridrato de Ponatinibe foi de 22 horas e as condições de estado de equilíbrio estacionário são tipicamente obtidas dentro de 1 semana de dose contínua. Com a dose uma vez por dia, as exposições plasmáticas do Cloridrato de Ponatinibe aumentam em cerca de 1,5 vezes entre a primeira dose e condições de estado de equilíbrio estacionário. Embora as exposições plasmáticas do Cloridrato de Ponatinibe aumentaram para níveis de estado de equilíbrio estacionário com a dose contínua, uma análise farmacocinética populacional prevê um aumento limitado na depuração oral aparente nas primeiras duas semanas de dose contínua, que não é considerada clinicamente relevante. O Cloridrato de Ponatinibe é principalmente eliminado através das fezes. Após dose oral única de Cloridrato de Ponatinibe com marcador [14C], aproximadamente 87% da dose radioativa é recuperada nas fezes e cerca de 5% na urina. O Cloridrato de Ponatinibe inalterado representou 24% e < 1%, da dose administrada, nas fezes e urina, respectivamente, com o restante da dose composta por metabólitos.
Insuficiência renal
Cloridrato de Ponatinibe não foi estudado em pacientes com insuficiência renal. Embora a excreção renal não seja uma via principal de eliminação do Cloridrato de Ponatinibe, a possibilidade de insuficiência renal moderado ou grave afetar a eliminação hepática não foi determinada.
Insuficiência hepática
Foi administrada uma dose única de 30 mg de Cloridrato de Ponatinibe a pacientes com insuficiência hepática leve, moderada ou grave e a voluntários saudáveis com função hepática normal. A Cmáx do Cloridrato de Ponatinibe foi comparável em pacientes com insuficiência hepática leve e em voluntários saudáveis com função hepática normal. Nos pacientes com insuficiência hepática moderada ou grave, a Cmáx e a AUC0-∞ do Cloridrato de Ponatinibe foram inferiores e a meia vida de eliminação plasmática do Cloridrato de Ponatinibe foi mais prolongada em pacientes com insuficiência hepática leve, moderada e grave, mas não significativamente diferente em termos clínicos de voluntários saudáveis com função hepática normal.
Os dados in vitro não demonstraram diferença na ligação às proteínas plasmáticas em participantes saudáveis e participantes com insuficiência hepática (leve, moderada e grave). Em comparação com voluntários saudáveis com função hepática normal, não foram observadas grandes diferenças na farmacocinética do Cloridrato de Ponatinibe em pacientes com diversos graus de insuficiência hepática. Não é necessária uma redução da dose inicial de Cloridrato de Ponatinibe em pacientes com insuficiência hepática.
Recomenda-se cuidado na administração de Cloridrato de Ponatinibe em pacientes com insuficiência hepática.
O Cloridrato de Ponatinibe não foi estudado em doses acima de 30 mg em pacientes com insuficiência hepática (Childs-Pugh Classes A, B e C)
Fatores intrínsecos que afetam a farmacocinética do Cloridrato de Ponatinibe
Não foram realizados estudos específicos para avaliar os efeitos do sexo, da idade, da raça e do peso corporal na farmacocinética do Cloridrato de Ponatinibe. Uma análise farmacocinética populacional integrada concluída relativa ao Cloridrato de Ponatinibe sugere que a idade pode ser preditiva da variabilidade da depuração oral aparente (CL/F) do Cloridrato de Ponatinibe. O sexo, a raça e o peso corporal não foram preditivos na explicação da variabilidade farmacocinética do Cloridrato de Ponatinibe entre os participantes.
Dados de segurança pré-clínica
Cloridrato de Ponatinibe foi avaliado em estudos de segurança farmacológica, toxicidade de dose repetida, genotoxicidade, toxicidade reprodutiva, fototoxicidade e carcinogenicidade.
O Cloridrato de Ponatinibe não exibiu propriedades genotóxicas quando avaliado nos sistemas padrão in vitro e in vivo.
As reações adversas não observadas durante os estudos clínicos, mas constatadas em animais sujeitos a níveis de exposição análogos aos níveis de exposição clínica, e com eventual relevância para a utilização clínica, são descritas a seguir.
Observou-se depleção dos órgãos linfoides em estudos de toxicidade de dose repetida em ratos e macacos cynomolgus. Os efeitos revelaram-se reversíveis após suspensão do tratamento.
Observaram-se alterações hiper/hipoplásticas dos condrócitos na placa de crescimento em estudos de toxicidade de dose repetida em ratos.
Em ratos, foram descobertas alterações inflamatórias acompanhadas por aumentos nos neutrófilos, monócitos, eosinófilos e nos níveis de fibrinogênios nas glandes do prepúcio e do clitóris após dosagem crônica.
Observaram-se alterações cutâneas sob a forma de crostas, hiperqueratose ou eritema em estudos de toxicidade em macacos cynomolgus. Observou-se pele seca e escamosa em estudos de toxicidade em ratos.
Em um estudo em ratos, observou-se edema difuso da córnea com infiltração de células neutrofílicas, e alterações hiperplásticas no epitélio lenticular sugestivos de uma reação fototóxica leve em animais tratados com 5 e 10 mg/kg de Cloridrato de Ponatinibe.
Em macacos cynomolgus, foram observados sopros cardíacos sistólicos sem correlações macroscópicas ou microscópicas em animais individuais tratados com 5 e 45 mg/kg no estudo de toxicidade de dose única e a 1, 2,5 e 5 mg/kg no estudo de toxicidade de dose repetida de 4 semanas. Desconhece-se a relevância clínica destes resultados.
Em macacos cynomolgus, observou-se atrofia folicular da glândula tiróide na maior parte dos casos acompanhada por uma redução nos níveis de T3 e uma tendência para níveis TSH aumentados no estudo de toxicidade de dose repetida de 4 semanas em macacos cynomolgus.
Observaram-se achados microscópicos relacionados ao Cloridrato de Ponatinibe nos ovários (atresia folicular aumentada) e testículos (degeneração mínima das células germinais) em animais tratados com 5 mg/kg de Cloridrato de Ponatinibe em estudos de toxicidade de dose repetida em macacos cynomolgus. Cloridrato de Ponatinibe em doses de 3, 10, e 30 mg/kg produziu aumentos no débito de urina e excreções de eletrólitos e provocou uma diminuição no esvaziamento gástrico em estudos de segurança farmacológica em ratos.
Em ratos, observou-se toxicidade embrio-fetal sob a forma de perda pós-implantação, peso fetal reduzido e múltiplas alterações dos tecidos moles e esqueleto em doses tóxicas maternas. Foram também observadas múltiplas alterações dos tecidos moles e esqueleto em doses maternas não tóxicas.
Em um estudo de fertilidade em ratos machos e fêmeas, verificou-se que, para níveis de dose correspondentes às exposições clínicas humanas, os parâmetros de fertilidade femininos foram reduzidos.
Foi relatada evidência de perda de embriões pré e pós implantação em ratos fêmeas e, portanto, o Cloridrato de Ponatinibe poderá comprometer a fertilidade feminina. Não foram observados efeitos nos parâmetros de fertilidade dos ratos machos. A relevância clínica destes resultados na fertilidade humana é desconhecida.
Observou-se, em ratos jovens, mortalidade relacionada com efeitos inflamatórios em animais tratados com 3 mg/kg/dia e observaram-se reduções do aumento de peso corporal com doses de 0,75, 1,5 e 3 mg/kg/dia durante as fases de tratamento pré-desmame e pós-desmame inicial. O Cloridrato de Ponatinibe não afetou de forma adversa importantes parâmetros de desenvolvimento no estudo de toxicidade juvenil.
Em um estudo de carcinogenicidade de dois anos em ratos machos e fêmeas, a administração oral de Cloridrato de Ponatinibe a 0,05, 0,1 e 0,2 mg/kg/dia em machos e a 0,2 e 0,4 mg/kg/dia em fêmeas não resultou em quaisquer efeitos tumorigênicos. A dose de 0,8 mg/kg/dia em fêmeas resultou num nível de exposição plasmática geralmente inferior ou equivalente à exposição humana no intervalo de dose de 15 mg a 45 mg diariamente. Foi observado um aumento estatisticamente significativo da incidência de carcinoma de células escamosas da glândula clitoridiana nessa dose. A relevância clínica deste resultado nos humanos é desconhecida.