 Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)
Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)Qual a ação da substância do Xospata?
Resultados de Eficácia
Eficácia e segurança clínicas
LMA recidivada ou refratária
A eficácia e a segurança foram avaliadas no estudo de fase 3 com controle ativo (2215- CL-0301) e dois estudos secundários de fase 1/2 com aumento da dose (2215-CL-0101 e 2215-CL-0102).
Estudo ADMIRAL (2215-CL-0301)
O estudo ADMIRAL é um estudo clínico de Fase 3, aberto, multicêntrico e randomizado de pacientes adultos com LMA recidivada ou refratária com mutação do FLT3 identificada por teste diagnóstico. Neste estudo 371 pacientes foram randomizados em uma proporção de 2:1 para receber gilteritinibe ou uma das seguintes quimioterapias de resgate (247 no grupo de gilteritinibe e 124 no grupo de quimioterapia de resgate).
- Citarabina 20 mg duas vezes ao dia por via subcutânea (SC) ou intravenosa (IV) durante 10 dias (dias 1 a 10) (LoDAC);
- Azacitidina 75 mg/m2 uma vez ao dia por via SC ou IV durante 7 dias (dias 1 a 7);
- Mitoxantrona 8 mg/m2 , etoposídeo 100 mg/m2 e citarabina 1000 mg/m2 uma vez ao dia por via IV durante 5 dias (dias 1 a 5) (MEC);
- Fator estimulador de colônias de granulócitos 300 mcg/m2 uma vez ao dia por via SC durante 5 dias (dias 1 a 5), fludarabina 30 mg/m2 uma vez ao dia por via IV durante 5 dias (dias 2 a 6), citarabina 2000 mg/m2 uma vez ao dia por via IV durante 5 dias (dias 2 a 6) e idarrubicina 10 mg/m2 uma vez ao dia por via IV durante 3 dias (dias 2 a 4) (FLAG-Ida).
Os pacientes incluídos eram recidivados ou refratários após a terapia de primeira linha para a LMA e foram estratificados de acordo com a resposta ao tratamento prévio para LMA e quimioterapia pré-selecionada, ou seja, de alta ou baixa intensidade. Embora o estudo incluísse pacientes com várias anormalidades citogenéticas relacionadas à LMA, foram excluídos pacientes com leucemia promielocítica aguda (LPA) ou LMA relacionada à terapia.
O gilteritinibe foi administrado por via oral com uma dose inicial de 120 mg por dia até uma toxicidade inaceitável ou ausência de benefício clínico. Foram permitidas reduções da dose para 80 mg (24,4%) para controle de reações adversas e aumentos da dose para 200 mg (31,7%) para pacientes que não responderam à dose inicial de 120 mg.
Dos pacientes que foram pré-selecionados para receber quimioterapia de resgate, 60,5% foram randomizados para alta intensidade e 39,5% para baixa intensidade. MEC e FLAG-Ida (esquemas de alta intensidade) foram administrados por até dois ciclos, dependendo da resposta ao primeiro ciclo. LoDAC e azacitidina (esquemas de baixa intensidade) foram administrados em ciclos contínuos de 4 semanas até toxicidade inaceitável ou ausência de benefício clínico.
As características demográficas e os parâmetros basais estavam bem equilibrados entre os dois grupos do tratamento. A idade mediana na randomização foi de 62 anos (entre 20 e 84 anos) no grupo de gilteritinibe e de 62 anos (entre 19 e 85 anos) no grupo de quimioterapia de resgate. No estudo, 42% dos pacientes tinham 65 anos ou mais e 12% tinham 75 anos ou mais. Cinquenta e quatro por cento dos pacientes eram mulheres. A maioria dos pacientes no estudo era de brancos (59,3%); 27,5% de asiáticos, 5,7% de negros, 4% de outras raças e 3,5% de etnias desconhecidas. A maioria dos pacientes (83,8%) apresentou uma pontuação do status de desempenho ECOG igual a 0 ou 1. Os pacientes tiveram as seguintes mutações confirmadas: FLT3-ITD apenas (88,4%), FLT3-TKD apenas (8,4%) ou FLT3-ITD e FLT3-TKD (1,9%). Doze por cento dos pacientes receberam tratamento prévio com outro inibidor de FLT3. A maioria dos pacientes tinha LMA com risco citogenético intermediário (73%), 10% tinham risco desfavorável, 1,3% risco favorável e 15,6%, risco citogenético não classificado.
Antes do tratamento com gilteritinibe, 39,4% dos pacientes tinham LMA refratária primária e a maioria desses pacientes foi classificada como refratária após 1 ciclo de quimioterapia de indução, 19,7% tinham LMA recidivada após transplante de célulastronco hematopoiéticas (TCTH) alogênico e 41% tinham LMA recidivada sem TCTH alogênico.
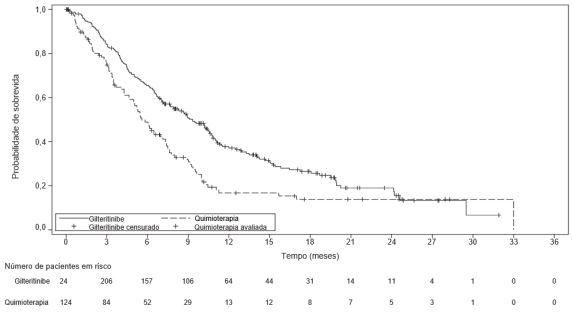
O desfecho primário de eficácia da análise final foi a sobrevida global (SG) da população ITT (intenção de tratar), mensurada desde a data da randomização até o óbito por qualquer causa (número de eventos analisados = 261). Os pacientes randomizados para o grupo de gilteritinibe apresentaram uma sobrevida consideravelmente mais longa, quando comparados ao grupo de quimioterapia (HR 0,637; IC de 95% 0,490– 0,830; valor de p unilateral: 0,0004. A sobrevida global mediana foi de 9,3 meses para os pacientes que estavam recebendo gilteritinibe e de 5,6 meses para os que estavam recebendo quimioterapia (Tabela 1, Figura 1).
Tabela 1. Estudo ADMIRAL - Sobrevida global em pacientes com LMA recidivada ou refratária
| Gilteritinibe (N=247) | Quimioterapia (N=124) | |
Óbitos, n (%) | 171 (69,2) | 90 (72,6) |
Mediana em meses (IC de 95%) | 9,3 (7,7, 10,7) | 5,6 (4,7, 7,3) |
Razão de risco (IC de 95%) | 0,637 (0,490, 0,830) | |
valor de p (unilateral) | 0,0004 | |
Taxa de sobrevida de 1 ano, % (IC de 95%) | 37,1 (30,7, 43,6) | 16,7 (9,9, 25) |
Figura 1. Método de Kaplan-Meier de sobrevida global no Estudo ADMIRAL
Uma análise modificada da sobrevida livre de eventos (SLE), definida como falha para a obtenção de remissão completa composta (RCc) com falhas designadas como um evento no Dia 1 do estudo, recidiva ou óbito decorrente de qualquer causa, incluindo eventos e início de novos tratamentos para leucemia relatados em um acompanhamento de longo prazo, mostrou uma melhora com mediana de 2,3 meses para o Hemifumarato de Gilteritinibe versus 0,7 mês para a quimioterapia de resgate HR 0,499 (IC de 95% 0,387, 0,643) e valor de p unilateral <0,0001.
A eficácia foi respaldada pela taxa de remissão completa (RC)/remissão completa com recuperação hematológica parcial (RCh), duração de RC/RCh (DOR) (Tabela 2) e taxa de conversão da dependência de transfusão para a independência de transfusão.
Tabela 2. Resultados de eficácia em pacientes com LMA recidivada ou refratária (Estudo ADMIRAL)
Taxa de remissão | Gilteritinibe (120 mg por dia) N=247 | Quimioterapia (N=124) |
RCa n/N (%) | 52/247 (21,1) | 13/124 (10,5) |
IC de 95%b | 16,1, 26,7 | 5,7, 17,3 |
DORc mediana (meses) | NR | 1,8 |
IC de 95%e | 11, NR | NE, NE |
RChd n/N (%) | 32/247 (13) | 6/124 (4,8) |
IC de 95%b | 9, 17,8 | 1,8, 10,2 |
DORc mediana (meses) | 4 | NE |
IC de 95%e | 2,1, 5,3 | NE, NE |
RC/RCh n/N (%) | 84/247 (34) | 19/124 (15,3) |
IC de 95%b | 28,1, 40,3 | 9,5, 22,9 |
DORc mediana (meses) | 11 | 1,8 |
IC de 95%e | 4,6, NR | NE, NE |
IC: intervalo de confiança; NE: não estimável
a. RC foi definida como contagem absoluta de neutrófilos ≥ 1,0 x 109 /L, plaquetas ≥ 100 x 109 /L, contagem diferencial normal da medula com < 5% de blastos, independência de transfusão de glóbulos vermelhos e plaquetas e nenhuma evidência de leucemia extramedular.
b. A taxa de IC de 95% foi calculada com o uso do método exato, com base na distribuição binomial.
c. DOR foi definida como o tempo desde a data da primeira RC ou RCh até a data de uma recidiva documentada de qualquer tipo.
d. RCh foi definida como blastos da medula < 5%, recuperação hematológica parcial, contagem absoluta de neutrófilos ≥ 0,5 x 109 /L e plaquetas ≥ 50 x 109/L, nenhuma evidência de leucemia extramedular e não poderia ter sido classificada como RC.
e. Com base nas estimativas de Kaplan-Meier.
Para os pacientes que obtiveram RC/RCh, o tempo mediano até a primeira resposta foi de 3,7 meses (intervalo: 0,9 a 10,6 meses) no grupo de gilteritinibe e de 1,2 mês (intervalo: 1 a 2,6 meses) no grupo da quimioterapia de resgate. O tempo mediano até a melhor resposta de RC/RCh foi de 3,8 meses (intervalo: 0,9 a 16 meses) no grupo de gilteritinibe e de 1,2 mês (intervalo: 1 a 2,6 meses) no grupo da quimioterapia de resgate.
Entre os 197 pacientes que eram dependentes de transfusões de glóbulos vermelhos e/ou plaquetas no período basal, 68 (34,5%) tornaram-se independentes de transfusões de glóbulos vermelhos e/ou plaquetas durante qualquer período de 56 dias pós-período basal. Para os 49 pacientes que eram independentes de transfusões de glóbulos vermelhos e/ou plaquetas no período basal, 29 (59,2%) continuaram independentes de transfusão durante qualquer período de 56 dias pós-período basal.
Estudo CHRYSALIS (2215-CL-0101)
O estudo CHRYSALIS é um estudo clínico aberto, multicêntrico e com escalonamento de dose que investiga a segurança, tolerabilidade, farmacocinética e farmacodinâmica da terapia com gilteritinibe em pacientes com LMA recidivada ou refratária com mutação no gene FLT3. As mutações no gene FLT3 foram identificadas por resultados locais.
Os pacientes foram incluídos em uma de sete coortes de escalonamento de dose ou expansão de dose, atribuídos para receber gilteritinibe uma vez ao dia em um dos níveis de doses a seguir: 20 mg, 40 mg, 80 mg, 120 mg, 200 mg, 300 mg ou 450 mg. Os pacientes foram avaliados quanto à expansão da coorte com base na segurança e tolerabilidade, inibição de FLT3 em estudos de correlação e atividade antileucêmica.
Neste estudo, 12 pacientes e 56 pacientes receberam gilteritinibe em um nível de dose diária de 80 mg e 120 mg, respectivamente. Os resultados de eficácia são apresentados de acordo com a dose aprovada.
No grupo de dose diária de 80 mg, a idade mediana dos pacientes era de 53 anos (intervalo: 21 a 71 anos), a maioria dos pacientes apresentava uma pontuação do status de desempenho ECOG de 0 ou 1 (58,3%) e a maioria dos pacientes apresentava mutações FLT3-ITD (83,3%); 8,3% apresentavam apenas a mutação FLT3-TKD e 8,3% apresentavam as mutações FLT3-ITD e FLT3-TKD. Dos pacientes com informações de raça, 75% eram caucasianos. No estudo, 58% eram do sexo feminino e 58,3% receberam três ou mais regimes antineoplásicos anteriores. No grupo de dose diária de 120 mg, a idade mediana dos pacientes era de 59 anos (intervalo: 23 a 87 anos), a maioria dos pacientes apresentava uma pontuação do status de desempenho ECOG de 0 ou 1 (73,2%) e a maioria dos pacientes apresentava mutações FLT3-ITD (83,9%); 10,7% apresentavam apenas a mutação FLT3-TKD e 5,4% apresentavam as mutações FLT3-ITD e FLT3-TKD. Dos pacientes com informações de raça, 87,5% eram caucasianos. No estudo, 54% eram do sexo feminino e 42,9% receberam três ou mais regimes antineoplásicos anteriores.
A eficácia foi respaldada pela taxa de remissão completa (RC)/remissão completa com recuperação hematológica parcial (RCh), duração de RC/RCh (DOR) e taxa de conversão da dependência de transfusão para a independência de transfusão. Os resultados de eficácia são apresentados na Tabela 3.
Tabela 3. Resultados de Eficácia em Pacientes com LMA Recidivada ou Refratária (Estudo CHRYSALIS)
Taxa de Remissão | Hemifumarato de Gilteritinibe (80 mg por dia) N=12 | Hemifumarato de Gilteritinibe (120 mg por dia) N=56 |
RCa n/N (%) | 2/12 (16,7) | 7/56 (12,5) |
IC de 95%b | 2,1, 48,4 | 5,2, 24,1 |
DOR medianac (meses) | NA | NR |
IC de 95%e | NA | 2,8, NE |
RChd n/N (%) | 1/12 (8,3) | 6/56 (10,7) |
IC de 95%b | 0,2, 38,5 | 4, 21,9 |
DOR medianac (meses) | NA | 2,1 |
IC de 95%e | NA | 0,6, 2,8 |
RC/RCh n/N (%) | 3/12 (25) | 13/56 (23,2) |
IC de 95%b | 5,5, 57,2 | 13, 36,4 |
DOR medianac (meses) | NA | 10,1 |
IC de 95%e | NA | 1,8, NE |
IC: intervalo de confiança; NE: não estimável; NR: não alcançado; NA: não disponível
a. RC foi definida como contagem absoluta de neutrófilos > 1,0 x 109 /L, plaquetas ≥ 100 x 109 /L, contagem diferencial normal da medula com < 5% de blastos, independência de transfusão de glóbulos vermelhos e plaquetas e nenhuma evidência de leucemia extramedular.
b. A taxa de IC de 95% foi calculada com o uso do método exato, com base na distribuição binomial.
c. DOR foi definida como o tempo desde a data da primeira RC ou RCh até a data de uma recidiva documentada de qualquer tipo.
d. RCh foi definida como blastos da medula < 5%, recuperação hematológica parcial, contagem absoluta de neutrófilos ≥ 0,5 x 109 /L e plaquetas ≥ 50 x 109 /L, nenhuma evidência de leucemia extramedular e não poderia ter sido classificada como RC.
e. Com base nas estimativas de Kaplan-Meier.
Para os pacientes que obtiveram RC/RCh, o tempo mediano até a primeira resposta de RC/RCh foi de 1,9 meses (intervalo: 0,9 a 4,6 meses) no grupo de 80 mg e 1,9 meses (intervalo: 1 a 9,2 meses) no grupo de 120 mg. O tempo mediano até a melhor resposta de RC/RCh foi de 1,9 meses (intervalo: 0,9 a 10,4 meses) no grupo de 80 mg e 2,1 meses (intervalo: 1 a 12 meses) no grupo de 120 mg.
Entre os 10 pacientes do grupo de 80 mg que eram dependentes de transfusões de glóbulos vermelhos e/ou plaquetas no período basal, 3 (30,0%) tornaram-se independentes de transfusões de glóbulos vermelhos e plaquetas durante qualquer período de 56 dias pós-período basal. Para os 2 pacientes que eram independentes de transfusões de glóbulos vermelhos e plaquetas no período basal, 1 (50,0%) continuou independente de transfusão durante qualquer período de 56 dias pós-período basal. De modo semelhante, entre os 52 pacientes do grupo de 120 mg que eram dependentes de transfusões de glóbulos vermelhos e/ou plaquetas no período basal, 11 (21,2%) tornaram-se independentes de transfusões de glóbulos vermelhos e plaquetas durante qualquer período de 56 dias pós-período basal. Para os 4 pacientes que eram independentes de transfusões de glóbulos vermelhos e plaquetas no período basal, 3 (75,0%) continuaram independentes de transfusão durante qualquer período de 56 dias pós-período basal.
Referências bibliográficas
Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, openlabel, phase 1-2 study . Lancet Oncol. 2017; (1470-2045).
Perl AE, Martinelli G, Cortes JE, et al. Gilteritinib Significantly Prolongs Overall Survival in Patients With FLT3-Mutated (FLT3mut+) Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML): Results From the Phase 3 ADMIRAL Trial [abstract]. 2019 Abstract # CT184.
Características Farmacológicas
Grupo farmacoterapêutico: agentes antineoplásicos, inibidores da proteína quinase, código ATC: L01XE54.
Mecanismo de ação
O fumarato de gilteritinibe é um inibidor de FLT3 e AXL.
Gilteritinibe inibe a sinalização do receptor FLT3 e a proliferação em células com expressão exógena de FLT3, inclusive FLT3-ITD, FLT3-D835Y e FLT3-ITD-D835Y, além de induzir apoptose em células leucêmicas com expressão de FLT3-ITD.
Efeitos farmacodinâmicos
Nos pacientes com LMA recidivada ou refratária que estavam recebendo gilteritinibe 120 mg, uma inibição substancial (> 90%) de fosforilação de FLT3 foi rápida (até 24 horas após a primeira dose) e sustentada, conforme caracterizado por um ensaio da atividade inibitória plasmática (PIA) ex vivo.
Intervalo QT prolongado
Foi observado um aumento relacionado à concentração na alteração do valor basal do QTcF em todas as doses de gilteritinibe de 20 a 450 mg. A alteração média prevista em relação ao valor basal do QTcF na Cmáx média em estado de equilíbrio (282,0 ng/mL) na dose diária de 120 mg foi de 4,96 ms com um IC unilateral superior de 95% = 6,20 ms.
Propriedades farmacocinéticas
Absorção
Após a administração oral de gilteritinibe, picos de concentrações plasmáticas são observados em um tmáx mediano de cerca de 4 a 6 horas em voluntários sadios e pacientes com LMA recidivada ou refratária. O gilteritinibe sofre uma absorção de primeira ordem com uma taxa de absorção estimada (ka) de 0,43 h-1 e um tempo de latência de 0,34 hora, com base na modelagem farmacocinética populacional. A concentração máxima (Cmáx) mediana em estado de equilíbrio é de 282,0 ng/mL (CV% = 50,8) e a área sob a curva de concentração plasmática durante um intervalo de administração de 24 horas (AUC0-24) é de 6180 ng·h/mL (CV% = 46,4) após a dose de 120 mg de gilteritinibe uma vez ao dia.
Níveis plasmáticos em estado de equilíbrio são alcançados em até 15 dias de administração uma vez ao dia com acúmulo aproximado de 10 vezes.
Efeito dos alimentos
Em adultos saudáveis, a Cmáx e a AUC de gilteritinibe diminuíram cerca de 26% e menos de 10%, respectivamente, quando uma dose única de 40 mg de gilteritinibe foi coadministrada com uma refeição com alto teor de gordura, comparado à exposição à gilteritinibe em jejum. O tmáx mediano foi atrasado 2 horas quando gilteritinibe foi administrado com uma refeição com alto teor de gordura.
Distribuição
A estimativa populacional do volume de distribuição central e periférico foi de 1092 L e 1100 L, respectivamente. Esses dados indicam que o gilteritinibe distribui-se amplamente fora do plasma, o que pode indicar extensa distribuição tecidual. A ligação às proteínas plasmáticas in vivo em seres humanos é de aproximadamente 90% e o gilteritinibe liga-se principalmente à albumina.
Biotransformação
Com base em dados in vitro, o gilteritinibe é metabolizado principalmente via CYP3A4. Os principais metabólitos em seres humanos incluem M17 (formado via Ndesalquilação e oxidação), M16 e M10 (ambos formados por N-desalquilação) e foram observados em animais. Nenhum desses três metabólitos excedeu 10% da exposição global ao composto original. A atividade farmacológica dos metabólitos com relação aos receptores FLT3 e AXL é desconhecida.
Interações medicamentosas dos transportadores
Experimentos in vitro demonstraram que o gilteritinibe é um substrato da P-gp e pode inibir a BCRP e P-gp no intestino delgado, OCT1 no fígado e MATE1 no rim em concentrações clinicamente relevantes.
Eliminação
Após uma dose única de gilteritinibe marcado com [14C], o gilteritinibe é excretado principalmente nas fezes, com 64,5% da dose total recuperada nas fezes.
Aproximadamente 16,4% da dose total foram eliminados na urina como medicamento inalterado e metabólitos. As concentrações plasmáticas do gilteritinibe caíram de maneira biexponencial com uma estimativa média de meia-vida na população de 113 horas. A depuração aparente estimada (CL/F) com base no modelo farmacocinético populacional é de 14,85 L/h.
Linearidade/não linearidade
Em geral, o gilteritinibe apresentou farmacocinética linear e proporcional às doses após a administração de dose única e múltiplas doses que variaram de 20 a 450 mg em pacientes com LMA recidivada ou refratária.
Dados de segurança pré-clínicos
Farmacologia de segurança
Em ratos foram observados: diminuição do fluxo urinário na dose de 30 mg/kg e em doses mais altas e redução do volume de fezes na dose de 100 mg/kg. Em cães foram observados: presença de sangue oculto nas fezes na dose de 10 mg/kg e em doses mais altas, diminuição da concentração de cálcio no sangue na dose de 30 mg/kg e salivação e aumento seguido de diminuição da concentração de cálcio no sangue na dose de 100 mg/kg. Essas alterações foram observadas nos níveis de exposição do plasma semelhantes ou inferiores aos níveis de exposição clínica.
Toxicidade de doses repetidas
Nos estudos de toxicidade de doses repetidas em ratos e cães, os órgãos mais afetados pela toxicidade foram o trato gastrintestinal (hemorragia em cães), sistema linfohematopoiético (necrose linfocítica e hipocelularidade da medula óssea com alterações nos parâmetros hematológicos), olho (inflamação e opacidade do cristalino em ratos, alteração do fundo de olho em cães e vacuolização da retina), pulmão (pneumonia intersticial em ratos e inflamação em cães), rim (alterações do túbulo renal com reação positiva de sangue oculto na urina) e fígado (vacuolização dos hepatócitos), bexiga (vacuolização epitelial) e tecido epitelial (úlcera e inflamação). Em ratos foi também observada fosfolipidose no pulmão e rim. Essas alterações foram observadas nos níveis de exposição do plasma semelhantes ou inferiores aos níveis de exposição clínica. A reversibilidade da maioria das alterações foi indicada ao final do período de recuperação de 4 semanas.
Toxicidade reprodutiva
Em estudos do desenvolvimento embriofetal o gilteritinibe demonstrou interrupção do crescimento fetal e induziu mortes embriofetais e teratogenicidade em ratos em níveis de exposição semelhantes aos níveis de exposição clínica. A transferência placentária do gilteritinibe foi demonstrada no rato, resultando na transferência de radioatividade para o feto, semelhante àquela observada no plasma materno.
O gilteritinibe foi excretado no leite de ratas lactantes com concentrações superiores às do plasma materno. O gilteritinibe distribuiu-se por meio do leite materno a diferentes tecidos, exceto para o cérebro, de ratos lactentes.
Estudo de toxicidade em animais jovens
No estudo de toxicidade em ratos jovens, o nível mínimo de dose letal (2,5 mg/kg/dia) foi muito inferior ao dos ratos adultos (20 mg/kg/dia). O trato gastrintestinal foi identificado como um dos órgãos mais afetados, semelhante ao que ocorre em ratos adultos.
Populações especiais
Uma análise farmacocinética populacional foi realizada para avaliar o impacto de covariáveis intrínsecas e extrínsecas na exposição prevista do gilteritinibe em pacientes com LMA recidivada ou refratária. A análise das covariáveis indicou que a idade (20 a 90 anos) e o peso corporal (36 kg a 157 kg) foram estatisticamente significativos; entretanto, a alteração prevista na exposição ao gilteritinibe foi inferior a 2 vezes.
Comprometimento hepático
O efeito do comprometimento hepático na farmacocinética de gilteritinibe foi estudado em pacientes com comprometimento hepático leve (Child-Pugh Classe A) e moderado (Child-Pugh Classe B). Os resultados indicam que a exposição ao gilteritinibe não ligado a pacientes com comprometimento hepático leve ou moderado é comparável à observada em pacientes com função hepática normal. O efeito do comprometimento hepático leve [definido por NCI-ODWG] sobre a exposição ao gilteritinibe também foi avaliado usando o modelo farmacocinético populacional, e os resultados demonstram pouca diferença na exposição prevista ao gilteritinibe em estado de equilíbrio com relação a um paciente típico com LMA recidivada ou refratária e função hepática normal.
Não é necessário ajustar a dose para pacientes com comprometimento hepático leve (Child-Pugh Classe A) ou moderado (Child-Pugh Classe B). O gilteritinibe não foi estudado em pacientes com insuficiência hepática grave (Child-Pugh Classe C).
Comprometimento renal
Não foi realizado um estudo dedicado ao comprometimento renal para avaliar o efeito da insuficiência renal na farmacocinética do gilteritinibe. O efeito do comprometimento renal leve ou moderado foi avaliado usando-se um modelo farmacocinético populacional. A creatinina sérica, um marcador da função renal, foi identificada como uma covariável estatisticamente significativa. Contudo, o aumento previsto da exposição ao gilteritinibe foi inferior a 2 -vezes. Não é necessário ajustar a dose em pacientes com comprometimento renal leve ou moderado. O efeito do comprometimento renal grave na exposição ao gilteritinibe não foi investigado.