 Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)
Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)Qual a ação da substância do Teriflunomida Accord Farma?
Resultados de Eficácia
A eficácia de Teriflunomida foi demonstrada em dois estudos fase 3, controlados com placebo, em pacientes com as formas recorrentes da esclerose múltipla, e um estudo fase 3, controlado com placebo, em pacientes com esclerose múltipla recente (isto é, com o primeiro episódio clínico).
O estudo 1 (EFC6049/TEMSO), duplo cego, controlado com placebo, avaliou doses únicas diárias de teriflunomida 7 mg e 14 mg, em pacientes com formas recorrentes da esclerose múltipla (EMR) durante 108 semanas. Todos os pacientes apresentavam o diagnóstico definitivo de esclerose múltipla (EM), exibindo um curso clínico de recidivas com ou sem progressão, com pelo menos uma recidiva no ano anterior ao estudo ou, pelo menos duas recidivas nos dois anos anteriores ao estudo.
Os indivíduos não tinham recebido interferon-beta por pelo menos 4 meses, ou qualquer outra medicação para EM por pelo menos 6 meses antes de entrar no estudo, nem foi permitido o uso destes medicamentos durante o estudo. Avaliações neurológicas foram realizadas na triagem, a cada 12 semanas até a semana 108 e em visitas não agendadas por suspeita de recidiva. Ressonância magnética de imagem (MRI) foi realizada na triagem, e nas semanas 24, 48, 72 e 108. O desfecho primário foi a taxa anual de recidivas (ARR).
Um total de 1088 pacientes com EMR foram randomizados para receber 7 mg (n=366) ou 14 mg (n=259) de teriflunomida ou placebo (n=363). Na inclusão, os pacientes tinham uma pontuação ≤ 5,5 na Escala Expandida do Estado de Incapacidade (EDSS). A idade média da população do estudo foi de 37,9 anos, a duração média da doença foi de 5,33 anos, e a média do EDSS basal foi de 2,68.
Um total de 91,4% apresentava EM recorrente-remitente (EMRR) e 8,6% tinham a forma de EM progressiva com recidivas. O tempo médio com placebo foi de 631 dias, com 7 mg de Teriflunomida foi de 635 dias, e com 14 mg de Teriflunomida foi de 627 dias.
A ARR foi significativamente reduzida em pacientes tratados com 14 mg de Teriflunomida, comparado com os pacientes que receberam placebo (Tabela 1). Houve uma redução consistente na ARR, observada nos subgrupos definidos por sexo, idade, terapia anterior da EM, e atividade basal da doença.
O risco de progressão da incapacidade sustentada por 12 semanas (conforme avaliado pelo aumento de, pelo menos 1 ponto, do EDSS basal ≤ 5,5, ou o aumento de 0,5 ponto para aqueles com EDSS basal > 5,5) foi estatisticamente menor apenas no grupo da teriflunomida 14 mg comparado com o placebo (Tabela 1 e Figura 1).
O efeito da teriflunomida em diversas variáveis de imagem na Ressonância Magnética (MRI) foi avaliado, incluindo o volume total de lesões T2 e lesões T1 hipointensas. A mudança no volume total das lesões comparado com o basal foi significativamente menor no grupo 14 mg do que no grupo placebo. Pacientes tratados com teriflunomida tiveram, significativamente menos lesões realçadas por gadolínio por imagem ponderada T1 do que aqueles no grupo placebo (Tabela 1).
Tabela 1: Resultados clínicos e de MRI do Estudo EFC6049/TEMSO
| - | Teriflunomida 14 mg (N=358) | Placebo (N= 363) |
Desfechos Clínicos | ||
Taxa Anual de Recidivas (desfecho primário) | 0,369 (p=0,0005) | 0,539 |
Redução do risco relativo | 31% | - |
Porcentagem de pacientes que permaneceram sem recidivas na semana 108 | 56,5% (p=0,0030) | 45,6% |
Porcentagem de progressão da incapacidade na semana 108 | 20,2% (p=0,028) | 27,3% |
Razão de azar | 0,70 | - |
Redução do risco relativo | 30% | - |
Desfecho MRI | ||
Alteração média do volume total de lesões1 basal (mL) na semana 108 | 0,345 (p=0,0003)2 | 1,127 |
Porcentagem de alteração relativa ao placebo | 69% | - |
Número médio de lesões T1 realçadas por gadolínio por imagem | 0,261 (p<0,0001) | 1,331 |
Redução relativa | 80% | - |
1Volume total de lesões: soma das lesões T2 e T1 hipointensas, volume em mL.
2Valores de p baseados na raiz cúbica transformada dos dados para volume total de lesões.
Figura 1: Gráfico Kaplan-Meier do tempo de progressão da incapacidade sustentada por 12 semanas – população intenção de tratar
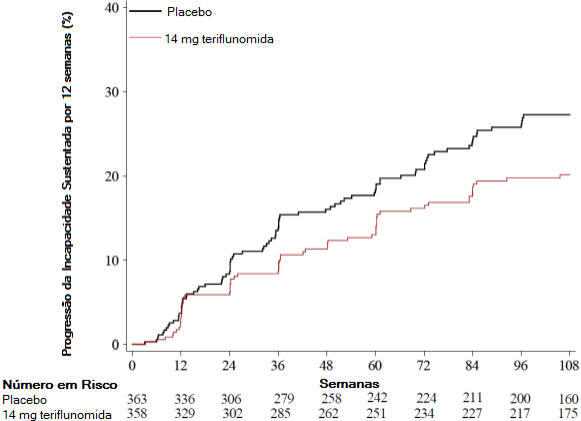
O estudo 2 (EFC10531/TOWER), duplo cego, controlado com placebo, avaliou doses únicas diárias de teriflunomida 7 mg e 14 mg em pacientes com as formas recorrentes da esclerose múltipla (EMR), com duração média do tratamento de, aproximadamente, 18 meses. Todos os pacientes tinham o diagnóstico definitivo de EM, exibindo um curso clínico de recidivas com ou sem progressão, com pelo menos uma recidiva durante o ano anterior ao estudo, ou pelo menos 2 recidivas durante os 2 anos anteriores ao estudo. Os indivíduos não haviam recebido interferon-beta, ou qualquer outro medicamento para EM, nos últimos 3 meses anteriores à inclusão no estudo, e nenhum destes medicamentos foi permitido durante o estudo. Avaliações neurológicas foram realizadas na triagem, a cada 12 semanas até o final do estudo, e em visitas não agendadas por suspeita de recidiva. O desfecho primário era a taxa anual de recidivas (ARR).
Um total de 1169 pacientes foram randomizados para receber 7 mg (n=408) ou 14 mg (n=372) de teriflunomida ou placebo (n=389). A idade média foi de 37,9 anos, a duração média da doença foi de 5,16 anos, e a média do EDSS basal foi de 2,7 (mediana do EDSS basal foi 2,50). A maioria dos pacientes apresentava EM recorrente-remitente (97,5%). O tempo médio com placebo foi de 571 dias, com Teriflunomida 7 mg foi de 552 dias, e com Teriflunomida 14 mg foi de 567 dias.
A ARR foi reduzida significativamente em pacientes tratados com 14 mg de Teriflunomida, comparado com os pacientes que receberam placebo (Tabela 2). Houve uma redução consistente na ARR, observada nos subgrupos definidos por sexo, idade, terapia anterior de EM, e atividade basal da doença.
O risco de progressão da incapacidade sustentada por 12 semanas (conforme avaliado pelo aumento de, pelo menos 1 ponto, do EDSS basal ≤ 5,5, ou o aumento de 0,5 ponto para aqueles com EDSS basal > 5,5) foi significantemente menor apenas no grupo de teriflunomida 14 mg comparado com placebo (Tabela 2 e Figura 2).
Tabela 2: Resultados clínicos do Estudo EFC10531/TOWER
| - | Teriflunomida 14 mg (N=370) | Placebo (N= 388) |
Desfechos Clínicos | ||
Taxa anual de recidivas (desfecho primário) | 0,319 (p=0,0001) | 0,501 |
Redução do risco relativo | 36,3% | - |
Porcentagem de pacientes que permaneceram sem recidivas na semana 108 | 57,1% (p<0,0001) | 46,8% |
Porcentagem da progressão da incapacidade na semana 108 | 15,8% (p=0,044) | 19,7% |
Razão de azar | 0,69 | - |
Redução do risco relativo | 31% | - |
Figura 2: Gráfico Kaplan-Meier do tempo para a progressão da incapacidade sustentada por 12 semanas – população com intenção de tratar
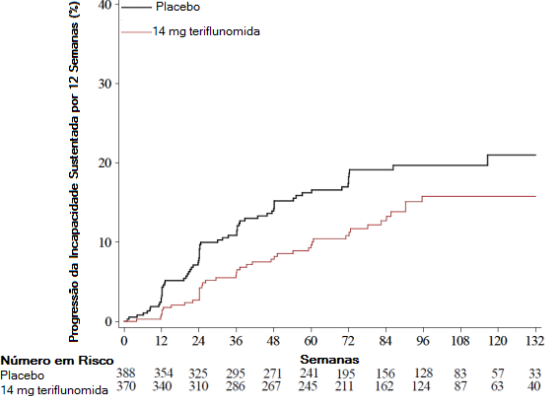
O Estudo 3 (EFC6260/TOPIC), duplo cego, controlado com placebo, avaliou doses únicas diárias de teriflunomida 7 mg e 14 mg por até 108 semanas em pacientes com EM recente (isto é, com o primeiro episódio clínico). Os pacientes apresentaram o primeiro evento neurológico num período de 90 dias da randomização, com 2 ou mais lesões T2, com pelo menos 3 mm de diâmetro que são característicos de EM. O desfecho primário foi o tempo para o segundo episódio clínico (recidiva).
Um total de 618 pacientes foram randomizados para receber 7 mg (n=205) ou 14 mg (n=216) de teriflunomida ou placebo (n=197). A idade média da população do estudo foi de 32,1 anos e o tempo médio desde o primeiro evento neurológico foi de 1,85 meses, 59,1% dos pacientes entraram no estudo com um episódio monofocal, e 40,9% com um episódio multifocal. O tempo médio com placebo foi de 464 dias, com Teriflunomida 7 mg foi de 464 dias, e com Teriflunomida 14 mg foi de 493 dias.
O risco do segundo episódio clínico foi reduzido de forma estatisticamente significativa no grupo de 14 mg de teriflunomida, comparado com o placebo (Tabela 3 e Figura 3).
O risco do segundo episódio clínico, ou uma nova lesão na MRI (uma nova lesão T1 ou T2 realçada por gadolínio), foi reduzida de forma estatística e significante no grupo de teriflunomida 14 mg, comparados com placebo (Tabela 3 e Figura 3).
Tabela 3: Resultados clínicos e de MRI do EFC6260/TOPIC
| - | Teriflunomida 14 mg (N=214) | Placebo (N= 197) |
Desfechos Clínicos | ||
Porcentagem de pacientes que permaneceram sem um segundo episódio clínico na semana 108 (desfecho primário) | 76,0% (p=0,0087) | 64,1% |
Razão de azar | 0,574 | - |
Porcentagem de pacientes que permaneceram sem um segundo episódio clínico e sem nova lesão MRI na semana 108 | 28,5% (p=0,0003) | 13,0% |
Razão de azar | 0,651 | - |
Desfecho MRI | ||
Alteração média do volume total de lesões1 basal (mL) na semana 108 | 0,227 (p=0,0374)2 | 0,202 |
Número médio de lesões T1 realçadas por gadolínio por imagem | 0,395 (p=0,0008) | 0,953 |
1Volume total de lesões: soma das lesões T2 e T1 hipointensas, volume em mL.
2Valores de p baseados na raiz cúbica transformada dos dados para volume total de lesões.
Figura 3: Gráfico Kaplan-Meier do tempo para o segundo episódio clínico – população com intenção de tratar (EFC6260/TOPIC)
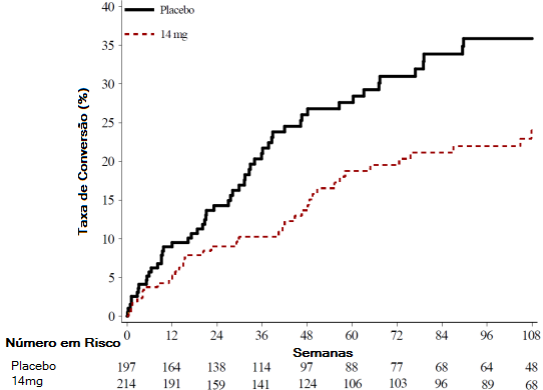
O efeito da teriflunomida na atividade MRI também foi demonstrada em um quarto estudo, randomizado, duplo cego, controlado com placebo, com pacientes com EM com recidivas. Um total de 179 pacientes foi tratado com o dobro da dose usual na primeira semana e, depois, recebeu 7 mg (n=61) ou 14 mg (n=57) de teriflunomida ou placebo (n=61) para o restante do período de 36 semanas de tratamento. O desfecho primário foi o número médio de lesões únicas ativas/MRI durante o tratamento. O exame RMI foi realizado no início, semana 6, semana 12, semana 18, semana 24, semana 30 e semana 36. O resultado demográfico basal foi consistente em todos os grupos de tratamento. O número médio de lesões ativas únicas por MRI do cérebro, durante o período das 36 semanas de tratamento, foi menor nos pacientes tratados com teriflunomida 14 mg (0,98) e 7 mg (1,06) quando comparado com placebo (2,69), sendo que a diferença foi estatisticamente significante para ambos (p=0,0052 e p=0,0234, respectivamente).
Referencias Bibliográficas
1. O'Connor P, Wolinsky JS, Confavreux C, Comi G, Kappos L, Olsson TP, Benzerdjeb H, Truffinet P, Wang L, Miller A, Freedman MS; TEMSO Trial Group. Randomized trial of oral teriflunomide for relapsing multiple sclerosis. N Engl J Med. 2011 Oct 6;365(14):1293-303.
2. Freedman MS; Teriflunomide in relapsing multiple sclerosis: therapeutic utility. Ther Adv Chronic Dis. 2013. Sep 4;4(5):192-205.
Características Farmacológicas
Farmacodinâmica
A teriflunomida é um agente imunomodulador com propriedades anti-inflamatórias que inibe de forma seletiva e reversível a enzima mitocondrial diidroorotato desidrogenase (DHO-DH), necessária para a síntese de novo de pirimidina. Como consequência, a teriflunomida bloqueia a proliferação dos linfócitos estimulados que necessitam da síntese de novo de pirimidina para expandir. O mecanismo exato pelo qual a teriflunomida exerce o seu efeito terapêutico na esclerose múltipla, não é totalmente conhecido, porém, pode incluir a redução do número de linfócitos ativados no sistema nervoso central. É provável que a teriflunomida diminua na periferia o número de linfócitos ativados disponíveis para migrarem para o sistema nervoso central.
Potencial para prolongar o intervalo QT
No estudo realizado com pacientes sadios controlados com placebo através de QT, a teriflunomida, em concentrações médias do estado de equilíbrio, não demonstrou qualquer potencial para prolongamento do intervalo QTcF comparado com o placebo: o maior tempo de diferença média pareado entre a teriflunomida e o placebo foi de 3,45 ms, com limite superior de IC 90% sendo 6,45 ms. Adicionalmente, nenhum valor de QTcF foi ≥ 480 ms e nenhuma alteração da linha basal foi > 60 ms.
Sistema imune
Efeito no número de células imunes no sangue – nos estudos controlados com placebo, 14 mg de teriflunomida uma vez ao dia levou à uma leve redução na contagem média de linfócitos, de menos de 0,3 x 109 /L, que ocorreu durante os três primeiros meses de tratamento, sendo que os níveis foram mantidos até o final do tratamento.
Em um estudo clínico, pacientes tratados com teriflunomida, apresentaram resposta imune apropriada à vacinação de gripe sazonal, consistente com a preservação de uma resposta à vacina de reforço. Pacientes dos grupos tratados com teriflunomida 7 mg e 14 mg atingiram títulos de anticorpos pós-vacinação consistentes com soroproteção: mais de 90% dos pacientes atingiram títulos de anticorpos pós-vacinação ≥ 40 para H1N1 e cepas B, em ambos os grupos de tratamento com teriflunomida. Para a cepa H3N2, títulos ≥ 40 foram atingidos em > 90% dos pacientes no grupo com 7 mg, e em 77% dos pacientes no grupo de 14 mg.
Em um segundo estudo de farmacodinâmica, a resposta imune à vacina inativada contra a raiva, neo antígeno, foi avaliada em um estudo duplo cego, randomizado, controlado com placebo, em indivíduos saudáveis. A média geométrica dos títulos para a vacina contra a raiva foi menor no grupo teriflunomida do que no grupo placebo, atingindo uma razão de tratamento pós-vacinação de teriflunomida versus placebo [IC 90%] de 0,53 [0,53; 0,81] no final da vacinação. Entretanto, após a vacinação, os níveis de anticorpos anti-raiva estavam acima de 0,5 UI/mL em todos os indivíduos, o limite para soroproteção. Neste mesmo estudo, a capacidade para montar uma reação de hipersensibilidade tipo tardia na pele para antígenos antigos como: Candida albicans, Trichophyton ou a Proteína Purificada Derivada da Tuberculina, em indivíduos recebendo teriflunomida, não diferiram do placebo.
Efeito nas funções renais tubulares
Nos estudos controlados com placebo, observou-se uma redução média do ácido úrico sérico de 20 a 30% em pacientes tratados com teriflunomida comparada com o placebo. A redução média de fósforo sérico foi de 10 a 15% no grupo de teriflunomida comparado com o placebo. Estes efeitos são considerados como relacionados ao aumento na excreção renal tubular e não são relacionadas às alterações nas funções glomerulares.
Farmacocinética
Baseado em uma análise farmacocinética populacional com teriflunomida utilizando dados de indivíduos saudáveis e pacientes com EM, t½ média, foi de aproximadamente 19 dias após doses repetidas de 14 mg. Leva-se aproximadamente 3 meses para atingir as concentrações de equilíbrio.
A razão área sob a curva (ASC) estimada de acúmulo é de aproximadamente 30, após doses repetidas de 14 mg.
Absorção
O tempo mediano para se atingir a concentração plasmática máxima ocorre entre 1 a 4 horas após a dose, seguido de administrações orais repetidas de teriflunomida, com alta biodisponibilidade (~100%).
Os alimentos não possuem efeito clínico relevante na farmacocinética da teriflunomida.
A exposição sistêmica aumenta de forma proporcional à dose, após a administração oral de 7 a 14 mg.
Distribuição
A teriflunomida se liga de forma extensiva à proteína plasmática (> 99%), e se distribui principalmente no plasma. O volume de distribuição é de 11 L após a administração intravenosa (IV) única.
Metabolismo
A teriflunomida é moderadamente metabolizada e grande parte circulante é detectada no plasma. A via primária de biotransformação para os metabólitos menores da teriflunomida é a hidrólise, sendo a oxidação uma via de menor importância. O caminho secundário envolve a oxidação, N-acetilação e conjugação com sulfato.
Eliminação
A teriflunomida é excretada no trato gastrintestinal principalmente através da bile, como droga inalterada, e possivelmente por secreção direta. Durante 21 dias, 60,1% da dose administrada são excretadas pelas fezes (37,5%) e urina (22,6%). Após o procedimento de eliminação acelerada com colestiramina, foram recuperadas 23,1% adicionais (principalmente nas fezes). Após a administração de dose única IV, o clearance corpóreo total da teriflunomida é de 30,5 mL/h.
Procedimento de eliminação acelerada - colestiramina e carvão ativado
A teriflunomida é eliminada do plasma lentamente. Sem o procedimento de eliminação acelerada, o tempo médio para a concentração plasmática atingir valores menores que 0,25 mg/L é de 6 meses. Devido às variações individuais no clearance do medicamento, este pode demorar até 2 anos. O procedimento de eliminação acelerada pode ser utilizado a qualquer momento após a descontinuação de Teriflunomida.
A eliminação pode ser acelerada pelos seguintes processos:
- Administração de 8 g de colestiramina, a cada 8 horas, por 11 dias. Se 8 g de colestiramina, três vezes por dia, não são bem tolerados, 4 g de colestiramina, três vezes por dia, podem ser usados;
- Administração de 50 g de pó de carvão ativado, a cada 12 horas, por 11 dias.
Se ambos os procedimentos de eliminação são pouco tolerados, os dias de tratamento não precisam ser consecutivos, a menos que seja necessário diminuir a concentração plasmática de teriflunomida rapidamente.
Ao final dos 11 dias, ambos esquemas aceleram a eliminação de teriflunomida com sucesso, levando à diminuição das concentrações plasmáticas de teriflunomida em mais de 98%.
O uso do procedimento de eliminação acelerada pode, potencialmente, resultar no retorno da atividade da doença, caso o paciente estiver respondendo ao tratamento com Teriflunomida.
Populações especiais
Sexo, idosos e crianças
Foram identificadas diversas fontes de variabilidade intrínseca em indivíduos saudáveis e em pacientes com esclerose múltipla, baseada na análise de farmacocinética populacional: idade, peso corpóreo, sexo, raça e níveis de albumina e de bilirrubina. No entanto, o impacto permanece limitado (≤ 31%).
Insuficiência hepática
A insuficiência hepática leve e moderada não teve impacto na farmacocinética da teriflunomida. A farmacocinética da teriflunomida em pacientes com insuficiência hepática severa, não foi avaliada.
Insuficiência renal
A insuficiência renal grave não teve impacto na farmacocinética da teriflunomida.