 Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)
Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)Qual a ação da substância do Ocrevus?
Resultados de Eficácia
Formas Recorrentes da Esclerose Múltipla (EMR)
A eficácia de Ocrelizumabe foi demonstrada em dois estudos clínicos randomizados, duplo-cegos, duplo-mascarados, controlados por comparador ativo de desenho idêntico, em pacientes com EMR tratados por 96 semanas (Estudo 1 e Estudo 2). A dose de Ocrelizumabe foi de 600 mg a cada 24 semanas (o tratamento inicial foi administrado com duas infusões intravenosas de 300 mg com um intervalo de 2 semanas, e as doses subsequentes foram administradas como uma infusão intravenosa única de 600 mg) e as injeções subcutâneas de placebo foram administradas 3 vezes por semana. A dose de REBIF, o comparador ativo, era de 44 mcg administrado como injeções subcutâneas 3 vezes por semana e as infusões intravenosas de placebo foram administradas a cada 24 semanas. Ambos os estudos incluíam pacientes que tinham apresentado pelo menos uma recidiva no ano anterior, ou duas recidivas em dois anos anteriores, e apresentavam uma pontuação na Escala Expandida do Estado de Incapacidade (Expanded Disability Status Scale - EDSS) de 0 a 5,5. Pacientes com formas primárias progressivas da esclerose múltipla (EM) foram excluídos. Avaliações neurológicas foram realizadas a cada 12 semanas e no momento de uma suspeita de recidiva. RMNs (Ressonância Magnética Nuclear) do cérebro foram realizadas na linha de base e nas semanas 24, 48 e 96.
O resultado primário tanto do Estudo 1 quanto do Estudo 2 foi a taxa de recidiva anual (TRA). Medidas de resultados adicionais incluíam a proporção de pacientes com progressão de incapacidade confirmada, o número médio de lesões realçadas por gadolínio (Gd) em RMN T1 nas semanas 24, 48 e 96, e lesões hiperintensas novas ou em aumento em RMN T2. A progressão de incapacidade foi definida como um aumento de 1 ponto ou mais desde a pontuação de EDSS basal atribuível à EM quando a pontuação de EDSS basal era de 5,5 ou menos, ou 0,5 pontos ou mais quando a pontuação de EDSS basal estava acima de 5,5. A progressão de incapacidade foi considerada confirmada quando o aumento na EDSS foi confirmado em uma visita regularmente programada 12 semanas após a documentação inicial de piora neurológica. A população primária para análise de progressão de incapacidade confirmada foi a população agrupada dos Estudos 1 e 2. No Estudo 1, 410 pacientes foram randomizados para Ocrelizumabe e 411 para REBIF. 11% dos pacientes tratados com Ocrelizumabe e 17% dos pacientes tratados com REBIF não concluíram o período de tratamento duplo-cego de 96 semanas. Os dados demográficos e características da doença na linha de base foram equilibrados entre os dois grupos de tratamento. Na linha de base, a idade média dos pacientes era de 37 anos; 66% eram mulheres. O tempo médio desde o diagnóstico da EM até a randomização foi de 3,8 anos, o número médio de recidivas no ano anterior foi de 1,3, e a pontuação média de EDSS foi de 2,8; 74% dos pacientes não tinham sido tratados com uma terapia não-esteroide para EM nos 2 anos anteriores ao estudo. Na visita basal, 40% dos pacientes apresentavam uma ou mais lesões realçadas por Gd em T1 (média 1,8).
No Estudo 2, 417 pacientes foram randomizados para Ocrelizumabe e 418 para REBIF; 14% dos pacientes tratados com Ocrelizumabe e 23% dos pacientes tratados com REBIF não concluíram o período de tratamento duplo-cego de 96 semanas. Os dados demográficos e as características da doença na visita basal foram equilibrados entre os dois grupos de tratamento. Na linha de base, a idade média dos pacientes era de 37 anos; 66% eram mulheres. O tempo médio desde o diagnóstico de EM até a randomização foi de 4,1 anos, o número médio de recidivas no ano anterior foi de 1,3, e a pontuação média na EDSS foi de 2,8; 74% dos pacientes não tinham sido tratados com uma terapia não-esteroide para EM nos 2 anos anteriores ao estudo. Na linha de base, 40% dos pacientes tratados com Ocrevus apresentavam uma ou mais lesões realçadas por Gd em T1 (média 1,9).
No Estudo 1 e no Estudo 2, Ocrelizumabe reduziu significativamente a taxa de recidiva anual e a proporção de pacientes com progressão de incapacidade confirmada em 12 semanas após o início em comparação com REBIF. Os resultados do Estudo 1 e do Estudo 2 são apresentados na Tabela 1 e na Figura 1.
Tabela 1: Principais Desfechos Clínicos e de RMN em Pacientes com EMR do Estudo 1 e do Estudo 2
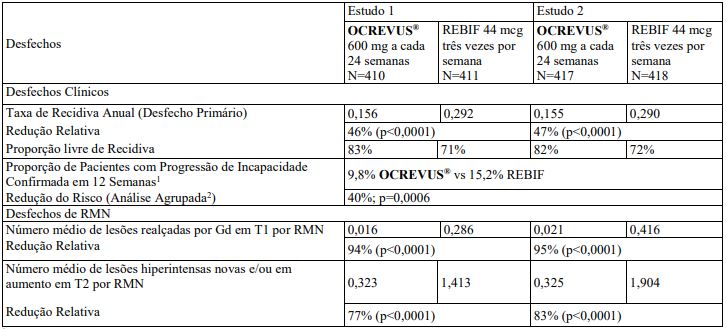
1 Definida como um aumento de 1,0 ponto ou mais da pontuação basal da Escala Expandida do Estado de Incapacidade (Expanded Disability Status Scale - EDSS) para pacientes com pontuação basal de 5,5 ou menos, ou 0,5 ou mais quando a pontuação basal é maior que 5,5, Estimativas de Kaplan-Meier na Semana 96.
2 Dados agrupados prospectivamente a partir do Estudo 1 e do Estudo 2.
Figura 1: Gráfico de Kaplan-Meier* do Tempo até o Início da Progressão de Incapacidade Confirmada Mantida por pelo menos 12 Semanas com o Evento Inicial de Agravamento Neurológico tendo ocorrido durante o Período de Tratamento Duplo-cego nos Estudos Agrupados 1 e 2 em Pacientes com EMR (População ITT Agrupada)
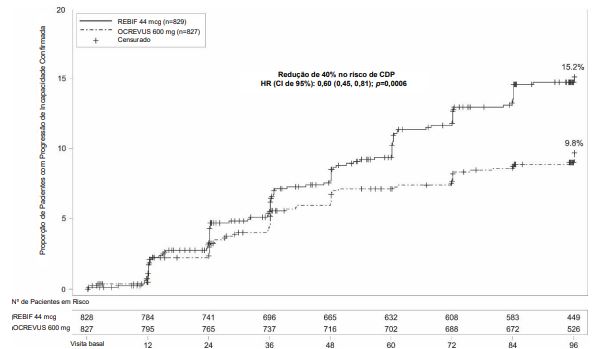
*Análise agrupada pré-especificada dos Estudos 1 e 2.
Em análises do subgrupo exploratório do Estudo 1 e do Estudo 2, o efeito de Ocrelizumabe na taxa de recidiva anual e na progressão de incapacidade foi semelhante em pacientes homens e mulheres.
Esclerose Múltipla Primária Progressiva (EMPP)
O Estudo 3 foi um estudo clínico randomizado, duplo-cego, controlado por placebo em pacientes com EMPP. Os pacientes foram randomizados a 2:1 para receber Ocrelizumabe 600 mg ou placebo como duas infusões intravenosas de 300 mg com um intervalo de 2 semanas a cada 24 semanas por pelo menos 120 semanas. Os critérios de seleção exigiam uma EDSS basal de 3 a 6,5 e uma pontuação de 2 ou mais para o sistema funcional piramidal da EDSS em razão de achados na extremidade inferior. Avaliações neurológicas foram conduzidas a cada 12 semanas. Uma RMN foi realizada na visita basal e nas Semanas 24, 48 e 120.
No Estudo 3, o resultado primário foi o tempo até o início da progressão de incapacidade atribuível à EM confirmada como presente na próxima avaliação neurológica pelo menos 12 semanas depois. A progressão de incapacidade ocorria quando a pontuação da EDSS aumentava 1 ponto ou mais a partir da EDSS basal se a EDSS basal fosse de 5,5 pontos ou menos, ou por 0,5 pontos ou mais se a EDSS basal fosse maior que 5,5 pontos. No Estudo 3, também foi considerado que a progressão de incapacidade confirmada tinha ocorrido se os pacientes que apresentassem progressão de incapacidade descontinuassem a participação no estudo antes da próxima avaliação. Medidas de resultado adicionais incluíam caminhada cronometrada por uma distância de 25 pés, e alteração percentual no volume da lesão hiperintensa em T2.
O Estudo 3 randomizou 488 pacientes para Ocrelizumabe e 244 para placebo; 21% dos pacientes tratados com Ocrelizumabe e 34% dos pacientes tratados com placebo não concluíram o estudo. Os dados demográficos e as características da doença na linha de base foram equilibrados entre os dois grupos de tratamento. Na linha de base, a idade média dos pacientes era de 45; 49% eram mulheres. O tempo médio desde o aparecimento do sintoma foi de 6,7 anos, a pontuação média na EDSS foi de 4,7, e 26% apresentaram uma ou mais lesões realçadas por Gd em T1 na visita basal; 88% dos pacientes não tinham sido tratados anteriormente com um tratamento não-esteroide para EM. O tempo até o início da progressão de incapacidade confirmada em 12 semanas após o início foi significativamente mais longo para pacientes tratados com Ocrelizumabe do que para pacientes tratados com placebo (vide Figura 2). Os resultados do Estudo 3 são apresentados na Tabela 2 e na Figura 2.
Tabela 2: Principais Desfechos Clínicos e de RMN em pacientes com EMPP no Estudo 3
Desfechos | Estudo 3 | |
| Ocrelizumabe 600 mg (duas infusões de 300 mg com um intervalo de duas semanas a cada 24 semanas) N=488 | Placebo N=244 | |
Resultados Clínicos | ||
Proporção de Pacientes com Progressão de Incapacidade Confirmada em 12 Semanas1 Redução de risco
| 32,9% | 39,3% |
24%; p=0,0321 | ||
Desfechos de RMN | ||
Alteração média no volume das lesões T2, desde a visita basal até a Semana 120 (cm3) | -0,39 | 0,79 |
p<0,0001 | ||
1 Definida como um aumento de 1,0 ponto ou mais a partir da pontuação de EDSS basal para pacientes com pontuação basal de 5,5 ou menos, ou um aumento de 0,5 ou mais quando a pontuação basal é maior que 5,5.
Figura 2: Gráfico de Kaplan-Meier do tempo até o início da progressão de incapacidade confirmada mantida por pelo menos 12 semanas com o evento inicial de agravamento neurológico tendo ocorrido durante o período de tratamento duplo-cego no Estudo 3*
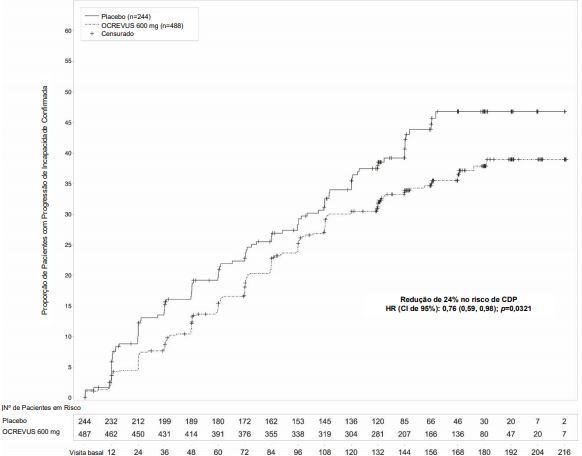
*Todos os pacientes nesta análise tinham no mínimo 120 semanas de acompanhamento. A análise primária baseia-se em todos os eventos de progressão de incapacidade acumulados, incluindo 21 sem EDSS confirmatória em 12 semanas.
Na população geral do Estudo 3, a proporção de pacientes com agravamento de 20 por cento na caminhada cronometrada de 25 pés confirmada em 12 semanas foi de 49% nos pacientes tratados com Ocrelizumabe em comparação com 59% nos pacientes tratados com placebo (redução de risco de 25%).
Nas análises do subgrupo exploratório do Estudo 3, a proporção de pacientes do sexo feminino com progressão de incapacidade confirmada em 12 semanas após o início foi semelhante nos pacientes tratados com Ocrelizumabe e nos pacientes tratados com placebo (aproximadamente 36% em cada grupo). Em pacientes do sexo masculino, a proporção de pacientes com progressão de incapacidade confirmada em 12 semanas após o início foi de aproximadamente 30% nos pacientes tratados com Ocrelizumabe e 43% nos pacientes tratados com placebo. Os desfechos clínicos e de RMN que geralmente favoreciam Ocrelizumabe numericamente na população geral, e que mostravam tendências semelhantes tanto em pacientes do sexo masculino quanto feminino, incluíam taxa de recidiva anual, alteração no volume da lesão em T2, e número de lesões novas ou em aumento em T2.
Características Farmacológicas
Farmacodinâmica Mecanismos de ação
O mecanismo preciso pelo qual ocrelizumabe exerce seus efeitos terapêuticos na esclerose múltipla é desconhecido, mas supõe-se que envolva a ligação ao CD20, um antígeno da superfície celular presente em linfócitos pré-B e linfócitos B maduros. Após a ligação da superfície celular aos linfócitos B, ocrelizumabe causa citólise celular dependente de anticorpo e lise mediada por complemento.
Propriedades farmacodinâmicas
Para contagens de células B, são usados ensaios de células B CD19+ pois a presença de Ocrelizumabe interfere no ensaio de CD20. O tratamento com Ocrelizumabe reduz as contagens de células B CD19+ no sangue 14 dias após a infusão. Em estudos clínicos, as contagens de células B aumentaram acima do limite inferior do normal (LIN) ou acima das contagens basais entre as infusões de Ocrelizumabe pelo menos uma vez em 0,3% a 4,1% dos pacientes. Em um estudo clínico de 51 pacientes, o tempo mediano para as contagens de células B voltarem ao valor basal ou LIN foi de 72 semanas (intervalo de 27-175 semanas) após a última infusão de Ocrelizumabe. Em 2,5 anos após a última infusão, as contagens de células B aumentaram até o valor basal ou LIN em 90% dos pacientes.
Propriedades farmacocinéticas
A farmacocinética (PK) de Ocrelizumabe em estudos clínicos de EM se enquadra em um modelo bicompartimental com a depuração dependente do tempo. A exposição geral no estado de equilíbrio (AUC durante os intervalos de dosagem de 24 semanas) de Ocrelizumabe foi de 3.510 mcg/mL por dia. Em estudos clínicos com pacientes com EM, as doses de manutenção de ocrelizumabe foram de 600 mg a cada 6 meses (pacientes com EMR) ou duas infusões de 300 mg separadas por um intervalo de 14 dias a cada 6 meses (pacientes com EMPP). A concentração máxima média foi de 212 mcg/mL para pacientes com EMR (infusão de 600 mg) e 141 mcg/mL para pacientes com EMPP (duas infusões de 300 mg administradas em duas semanas). A farmacocinética de ocrelizumabe foi essencialmente linear e proporcional à dose entre 400 mg e 2000 mg.
Absorção
Ocrelizumabe é administrado em infusão endovenosa. Não foram realizados estudos com outras formas de administração.
Distribuição
A estimativa de PK da população do volume de distribuição central foi de 2,78 L. O volume periférico e a depuração intercompartimental foram estimados em 2,68 L e 0,29 L/dia, respectivamente.
Metabolismo
O metabolismo de Ocrelizumabe não foi estudado diretamente, pois a depuração de anticorpos ocorre principalmente por catabolismo.
Eliminação
A depuração constante foi estimada em 0,17 L/dia, e a depuração inicial dependente do tempo em 0,05 L/dia, que diminuiu com uma meia-vida de 33 semanas. A meia-vida de eliminação terminal foi de 26 dias.
Farmacocinética em populações especiais
Pediatria
Não foram conduzidos estudos para investigar a farmacocinética de Ocrelizumabe em crianças e adolescentes (menos de 18 anos de idade).
Idosos
Não foram conduzidos estudos para investigar a farmacocinética de Ocrelizumabe em pacientes com 65 anos ou mais.
Insuficiência renal
Pacientes com comprometimento renal leve foram incluídos nos estudos clínicos. Nenhuma alteração significativa na farmacocinética de Ocrelizumabe foi observada nesses pacientes.
Insuficiência hepática
Pacientes com comprometimento hepático leve foram incluídos em estudos clínicos. Nenhuma alteração significativa na farmacocinética de Ocrelizumabe foi observada nesses pacientes.
Segurança pré-clínica
Carcinogênese, Mutagênese, Comprometimento da Fertilidade
Nenhum estudo de carcinogenicidade foi realizado para avaliar o potencial carcinogênico de Ocrelizumabe.
Nenhum estudo foi realizado para avaliar o potencial mutagênico de Ocrelizumabe. Como um anticorpo, não se espera que Ocrelizumabe interaja diretamente com o DNA.
Não foi observado nenhum efeito nos órgãos reprodutores em macacos machos que receberam ocrelizumabe por injeção intravenosa (três doses iniciais de 15 ou 75 mg/kg, seguidas por doses semanais de 20 ou 100 mg/kg) durante 8 semanas. Também não foi observado efeito no ciclo estral em macacas que receberam ocrelizumabe durante três ciclos menstruais usando o mesmo regime de dosagem. As doses testadas em macacos são 2 e 10 vezes a dose humana recomendada de 600 mg, baseado em mg/kg.
Teratogenicidade
Após a administração intravenosa de Ocrelizumabe a macacas durante a organogênese (doses iniciais de 15 ou 75 mg/kg nos dias de gestação 20, 21 e 22, seguidas por doses semanais de 20 ou 100 mg/kg), observou-se depleção de linfócitos B no tecido linfoide (baço e linfonodos) nos fetos em ambas as doses.
A administração intravenosa de Ocrelizumabe (três doses iniciais diárias de 15 ou 75 mg/kg, seguidas por doses semanais de 20 ou 100 mg/kg) a macacas prenhas durante todo o período de organogênese e continuando até o período neonatal resultou em mortes perinatais (algumas associadas a infecções bacterianas), toxicidade renal (glomerulopatia e inflamação), formação de folículo linfoide na medula óssea, e diminuições severas nos linfócitos B circulantes em recém-nascidos. A causa das mortes neonatais é incerta, porém, constatou-se que ambos os neonatos afetados apresentavam infecções bacterianas. Peso testicular reduzido foi observado nos recém-nascidos sob a dosagem alta.
Não foi identificada dose sem efeito quanto a efeitos adversos no desenvolvimento, as doses testadas em macacos são 2 e 10 vezes a dose recomendada em humanos de 600 mg, baseado em mg/kg.