 Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)
Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)Qual a ação da substância do Nerlynx?
Resultados de Eficácia
No estudo principal de Fase III, multicêntrico, randomizado, duplo cego, controlado com placebo, ExteNET (3004), 2840 mulheres com câncer de mama HER2 positivo em fase inicial (conforme confirmado localmente por exames) que concluíram o tratamento adjuvante com trastuzumabe foram randomizadas 1:1 para receber Maleato de Neratinibe ou placebo, todos os dias, durante um ano. O estudo foi divido em três partes sendo elas: Parte A – seguimento de 2 anos para a sobrevida livre de doença invasiva (invasive disease-free survival, iDFS), Parte B – seguimento de 5 anos para iDFS e Parte C – sobrevida em longo prazo. A média de idades na população «intenção de tratar» (ITT) era de 52 anos (59,9% tinham ≥ 50 anos, 12,3% tinham ≥ 65 anos); 81,0% eram caucasianos, 2,6% negros ou afro-americanos, 13,6% asiáticos e 2,9% outros. No início do estudo, 57,7% tinham doença com receptores hormonais positivos (definida como ER-positiva e/ou PgR-positiva), 27,2% tinham gânglios negativos, 41,5% tinham um a três gânglios positivos e 29,4% tinham quatro ou mais gânglios positivos. Aproximadamente 10 % dos pacientes tinham tumores no estágio I, aproximadamente 40% tinham tumores no estágio II e aproximadamente 30 % tinham tumores no estágio III. A maioria dos pacientes (81%) foi incluída no prazo de um ano após a conclusão do tratamento com trastuzumabe. O tempo mediano desde o último tratamento adjuvante com trastuzumabe até à randomização foi de 4,5 meses.
O endpoint primário do estudo foi a sobrevida livre de doença invasiva (invasive disease-free survival, iDFS), definida como o tempo entre a data da randomização até a primeira ocorrência da recidiva invasiva (câncer de mama local/regional, ipsilateral ou contralateral), recorrência à distância, ou morte por qualquer causa, com 2 anos e 28 dias de acompanhamento. Os endpoints secundários do estudo incluíram a sobrevida livre de doença (disease-free survival, DFS) incluindo carcinoma ductal in situ (DFS-DCIS), tempo até à recorrência à distância (time to distant recurrence, TTDR), sobrevida livre de doença à distância (distant disease-free survival, DDFS), incidência cumulativa da recorrência no sistema nervoso central e sobrevida global (overall survival, OS).
A análise primária do estudo 2 anos após a randomização demonstrou que Maleato de Neratinibe reduziu significativamente o risco de recorrência da doença invasiva ou morte em 33% ( hazard ratio=0,67 com IC 95% (0,49, 0,91), bilateral (p = 0,011)) na população ITT.
Tabela 1: Resultados de eficácia primária após 2 anos – populações ITT e populações com receptores hormonais positivos que tenham concluído o tratamento com trastuzumabe há menos de um ano.
| Variável | Taxas livres de eventos estimados a 2 anos1 (%) | Hazard ratio (IC 95%)2 | Valor de p3 | |
| População ITT | ||||
| Maleato de Neratinibe (N=1420) | Placebo (N=1420) | |||
| Sobrevida livre de doença invasiva | 94,2 | 91,9 | 0,66 (0,49; 0,91) | 0,011 |
| Sobrevida livre de doença, incluído carcinoma ductal in situ | 94,2 | 91,3 | 0,62 (0,46; 0,84) | 0,002 |
| Sobrevida livre de doença à distância | 95,3 | 94,0 | 0,75 (0,53;1,06) | 0,110 |
| Tempo até a recorrência à distância | 95,5 | 94,2 | 0,74 (0,52; 1,06) | 0,102 |
| Recorrência no SNC | 0,92 | 1,16 | - | 0,586 |
| População com receptores hormonais positivos que tenha concluído o tratamento com trastuzumabe há menos de um ano | ||||
| Maleato de Neratinibe (N=671) | Placebo (N=668) | Hazard ratio (IC 95%)4 | Valor de p5 | |
| Sobrevida livre de doença invasiva | 95,3 | 90,9 | 0,50 (0,31; 0,78) | 0,003 |
| Sobrevida livre de doença, incluído carcinoma ductal in situ | 95,3 | 90,1 | 0,45 (0,28; 0,71) | <0,001 |
| Sobrevida livre de doença à distância | 96,1 | 93,0 | 0,53 (0,31; 0,88) | 0,015 |
| Tempo até a recorrência à distância | 96,3 | 93,3 | 0,53 (0,30; 0,89) | 0,018 |
| Recorrência no SNC | 0,34 | 1,01 | - | 0,189 |
SNC = sistema nervoso central.
1 Taxas livres de eventos para todos os endpoints, exceto para a recorrência no SNC para a qual a incidência cumulativa é notificada.
2 Modelo de riscos proporcionais de Cox estratificado.
3 Teste log-rank bilateral estratificado para todos os endpoints, exceto para a recorrência no SNC, para a qual foi utilizado o método de Gray.
4 Modelo de riscos proporcionais de Cox não estratificado.
5 Teste log-rank bilateral não estratificado para todos os endpoints, exceto para a recorrência do SNC, para a qual foi utilizado o método de Gray.
Figura 1: Diagrama de Kaplan-Meier da sobrevida livre de doença invasiva – População com receptores hormonais positivos que tenha concluído o tratamento com trastuzumabe há menos de um ano.
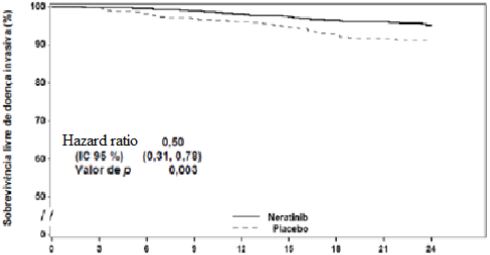
Número em risco | Meses após randomização | ||||||||
| Neratinibe | 671 | 606 | 593 | 577 | 559 | 539 | 517 | 486 | 309 |
| Placebo | 668 | 642 | 622 | 605 | 583 | 655 | 544 | 504 | 327 |
Para os pacientes com receptores hormonais positivos, que tenham concluído o tratamento com transtuzumabe há menos de um ano, o benefício relativo do tratamento com Maleato de Neratinibe em subgrupos de pacientes pré-estratificados é apresentado na Figura 2.
Figura 2: Sobrevida livre de doença invasiva em pacientes com receptores hormonais positivos que tenham concluído a terapia com transtuzumabe há menos de um ano, por subgrupo de pacientes.
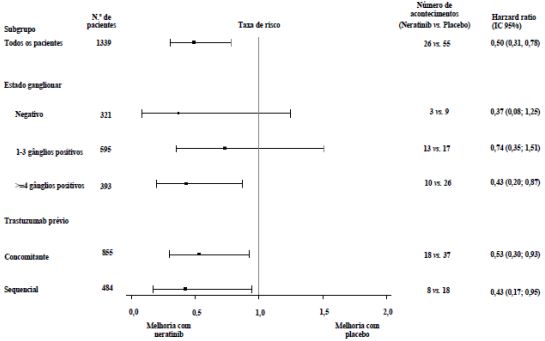
Nota: Pacientes (n=30) com estado ganglionar desconhecido não se encontram representados, uma vez que a taxa de risco não pode ser estimada.
Nos pacientes com receptores hormonais negativos (n= 1.209 (42,57%)), independentemente da duração do tratamento com trastuzumabe, a hazard ratio para iDFS aos 2 anos foi de 0,94, com um IC 95% (0,61; 1,46). Nesta população, a eficácia não foi demonstrada.
Cerca de 75% dos pacientes foram aprovados para seguimento prolongado além dos 24 meses. As observações com dados faltantes foram excluídas na última data de avaliação. Embora o benefício terapêutico de Maleato de Neratinibe relativamente ao placebo se tivesse mantido em cinco anos, a dimensão do efeito não pode ser estimada de forma segura.
A mediana de seguimento da Sobrevida Global (SG) na população ITT foi de 8,06 anos, 8,03 anos no braço neratinibe e 8,10 anos no braço placebo, com um total de 1542 (54,3%) dos pacientes seguidos para uma sobrevida durante 8 ou mais anos, 746 (52,5%) no braço neratinibe e 796 (56,1%) no braço placebo. O número de mortes foi 264 (9,3%), com 127 (8,9%) dos pacientes tratados com neratinibe e 137 (9,6%) nos pacientes tratados com placebo.
Não houve diferença estatisticamente significativa na sobrevida global entre o braço Maleato de Neratinibe e o braço placebo [HR 0,96 (IC 95%: 0,75; 1,22)] na população ITT numa mediana de seguimento de 8,06 anos.
Na população com receptores hormonais positivos que tinha concluído o tratamento com trastuzumabe há menos de um ano, a mediana de seguimento foi de 8,0 anos no braço neratinibe e 8,1 anos no braço placebo, com um total de 1339 (47,1%) pacientes por 8 ou mais anos, 671 (23,6%) no braço neratinibe e 668 (23,5%) no braço placebo. Nesta subpopulação, o número de mortes foi 55 (8,2%) nos pacientes tratados com neratinibe e 68 (10,2%) nos pacientes tratados com placebo [HR 0,83 (IC 95%: 0,58; 1,18)].
Características Farmacológicas
Propriedades farmacodinâmicas
Mecanismo de ação
O neratinibe é um inibidor da tirosina quinase (TKI) homólogo do oncogene viral da leucemia paneritroblástica (ERBB) irreversível que bloqueia a transdução dos sinais do fator de crescimento mitogênico através da ligação covalente de alta afinidade ao local de ligação do ATP de 3 receptores do fator de crescimento epidérmico (EGFR): EGFR (codificado por ERBB1), HER2 (codificado por ERBB2) e HER4 (codificado por ERBB4) ou os seus heterodímeros ativos com HER3 (codificado por ERBB3). Isto resulta na inibição sustentada destas vias promotoras do crescimento do câncer de mama com sobre-expressão ou amplificação do HER2 ou câncer de mama com HER2 mutante. O neratinibe liga-se ao receptor HER2, reduz a auto fosforilação de EGFR e HER2, a jusante das vias de sinalização de MAPK e AKT, e inibe potencialmente a proliferação celular tumoral in vitro. O neratinibe inibiu a expressão de EGFR e/ou HER2 em linhagens celulares de carcinoma com um IC50 celular <100 nM.
Propriedades Farmacocinéticas
O balanço de massas após a administração de uma dose oral única de 200 mg de neratinibe foi estudado em seis indivíduos saudáveis.
Absorção
Após a administração oral de 240 mg de neratinibe, a absorção foi lenta e as concentrações plasmáticas máximas de neratinibe ocorreram aproximadamente 7 horas após a administração. Uma dose única de 240 mg de neratinibe tomada com alimentos aumentou a Cmáx e a AUC em aproximadamente 17% e 13%, respectivamente, em comparação com a administração em jejum. Uma dose oral única de 240 mg de neratinibe tomada com uma refeição rica em gorduras aumentou tanto a Cmáx como a AUC em aproximadamente 100%. Num estudo de balanço de massas, a recuperação total (excreção urinária e fecal) de neratinibe intacto e metabólitos demonstrou que a fração absorvida para o neratinibe é de pelo menos 10% e provavelmente superior a 20%. Adicionalmente, as estimativas baseadas em modelos sugerem uma fração total absorvida pelo intestino (fa) de 26%.
A solubilidade in vitro do neratinibe é dependente do pH. Os tratamentos que aumentam o pH gastrointestinal podem reduzir a absorção do neratinibe, diminuindo assim a exposição sistêmica.
Distribuição
A ligação do neratinibe às proteínas plasmáticas humanas, incluindo a ligação covalente à albumina sérica humana (HSA), foi superior a 98% e independente da concentração de neratinibe testada. O neratinibe ligase predominantemente à HSA e alfa-1-glicoproteína ácida humana (AAG). A ligação do metabolito principal M6 (M6) às proteínas plasmáticas humanas foi superior a 99% e independente das concentrações de M6 testadas.
Estudos in vitro demonstraram que o neratinibe é um substrato da glicoproteína-P (P-gp-gp) (veja itens 4. Contraindicações, 5. Advertências e Precauções e 8. Posologia e Modo de usar) e BCRP. Os estudos in vitro demonstraram que o neratinibe e o seu principal metabolito M6 não são substratos dos transportadores hepáticos de captação OATP1B1*1a e OATP1B3 a 10 μM.
Biotransformação
O neratinibe é metabolizado principalmente nos microssomas hepáticos pelo CYP3A4 e, até certo ponto, pela flavina monoxigenase (FMO).
O perfil preliminar do metabolito no plasma humano indica que, após administração oral, o neratinibe sofre um metabolismo oxidativo através do CYP3A4. Os metabolitos circulantes incluem neratinibe piridina Nóxido (M3), N- desmetil neratinib (M6), neratinib dimetilamina N-óxido (M7) e traços de hidroxil neratinib N-óxido e neratinib bis-N-óxido (M11). O neratinib representa o componente mais proeminente no plasma e entre os metabolitos circulantes (M2, M3, M6, M7 e M11) nenhum se encontra acima de 8% da exposição total ao neratinibe mais o metabólito após a administração oral de neratinibe. Os metabólitos do neratinibe M3, M6, M7 e M11 mostraram ter potências semelhantes ao neratinibe em ensaios in vitro, tanto em ensaios de ligação (através de enzimas) como em ensaios baseados em linhagens celulares expressando ERBB1, ERBB2 (HER2) e ERBB4.
Com base nas exposições no estado estacionário, o neratinibe proporciona a maior parte da atividade farmacológica (73%), com 20% proporcionada pela exposição ao M6, 6% proporcionada pelo M3 e uma contribuição negligenciável (<1%) da AUC do M7 e do M11.
Eliminação
Após a administração de doses únicas de neratinibe, a meia vida média aparente do neratinibe no plasma dos doentes foi de 17 horas.
O neratinibe é excretado principalmente através das fezes
Após a administração de uma única dose radiomarcada de 240 mg de solução oral de neratinibe, 95,5% e 0,96% da dose administrada foram recuperadas nas fezes e na urina, respectivamente.
A excreção foi rápida e completa, com a maior parte da dose recuperada nas fezes no período de 48 horas e 96,5% da radioatividade total recuperada nas fezes e urina após 8 dias.
O neratinibe inalterado foi a espécie mais abundante nas fezes e urina, responsável por 62,1% da dose total recuperada. O metabólito mais abundante nas fezes foi o M6 (19,7% da dose administrada), seguido do M2, M3 e M7, todos abaixo dos 10% da dose administrada.
Interações medicamentosas
Efeito dos indutores do CYP3A4/ P-gp sobre o neratinibe
Após a administração concomitante de 240 mg de neratinibe e doses repetidas de 600 mg de rifampicina, um indutor potente do CYP3A4/P-gp, as exposições ao neratinibe diminuíram significativamente em 76% e 87% em relação à Cmax e à AUC, respetivamente, em comparação com a administração de neratinibe isolado.
Efeito dos inibidores do CYP3A4/P-gp sobre o neratinibe
A administração concomitante de uma dose oral única de 240 mg de neratinibe na presença de cetoconazol (400 mg uma vez por dia durante 5 dias), um inibidor potente do CYP3A4, aumentou a exposição sistêmica ao neratinibe em 3,2 e 4,8 vezes em relação à Cmax e à AUC, respetivamente, em comparação com neratinibe isolado.
As estimativas baseadas em modelos sugerem que a administração concomitante de uma dose oral única de 240 mg de neratinibe na presença de fluconazol (200 mg uma vez por dia durante 8 dias), um inibidor moderado do CYP3A4/P-gp, aumentou a exposição sistémica ao neratinibe em 1,3 e 1,7 vezes em relação à Cmax e à AUC, em comparação com neratinibe isolado.
As estimativas baseadas em modelos sugerem que a administração concomitante de uma dose oral única de 240 mg de neratinibe na presença de verapamil (120 mg duas vezes por dia durante 8 dias), um inibidor moderado do CYP3A4/P-gp, aumentou a exposição sistêmica ao neratinibe em 3,0 e 4,0 vezes em relação à Cmax e à AUC, em comparação com neratinibe isolado (veja itens 5. Advertências e Precauções, 6. Interações Medicamentosas e 8. Posologia e modo de usar).
Efeito dos modificadores do pH gástrico sobre o neratinibe
A administração concomitante de lansoprazol ou ranitidina (1x300 mg) com uma dose única de 240 mg de neratinibe em voluntários saudáveis resultou numa diminuição da exposição ao neratinibe de cerca de 70% ou 50%, respetivamente. A magnitude da interação da ranitidina na AUC do neratinibe foi reduzida em cerca de 25%, com o escalonamento da administração da ranitidina (2x150 mg) 2 horas após a administração de neratinibe.
Efeito de outros tratamentos sobre o neratinibe
Não foram observadas interações medicamentosas clinicamente relevantes aparentes quando o neratinibe foi administrado concomitantemente com capecitabina, paclitaxel, trastuzumab, vinorelbina ou antidiarreicos (loperamida).
Efeito de neratinibe sobre os substratos CYP
O neratinibe e o metabólito M6 não foram inibidores potentes diretos do CYP1A2, 2A6, 2B6, 2C8, 2C9, 2D6 ou 3A4. A inibição dependente do tempo do CYP3A4 e do CYP2B6 pelo neratinibe e pelo M6 não pode ser excluída.
O neratinibe não induziu o CYP1A2, 2B6, 2C9 ou 3A4.
Efeito do neratinibe sobre os transportadores
Não foi observada inibição clinicamente relevante da atividade in vitro do transportador de efluxo BSEP (bile salt export pump) humano, com um valor de IC50 notificado > 10 μM. O neratinibe a 10 μM pareceu inibir o transportador de efluxo BCRP, o que poderá ser clinicamente relevante a nível intestinal.
Em estudos in vitro, o neratinibe foi um inibidor dos transportadores de efluxo da glicoproteína P (P-gp), o que foi também confirmado num estudo clínico. Múltiplas doses orais de neratinibe 240 mg aumentaram as exposições à digoxina (aumento de 54 e 32% na Cmax e AUC, respetivamente) sem impacto no nível de depuração renal.
O neratinibe não produziu qualquer atividade inibitória em relação aos transportadores de captação, OATP1B1*1a, OATP1B3, OAT1, OAT3 e OCT2, com valores de IC50 notificados > 10μM. O neratinibe produziu atividade inibitória no transportador de captação OCT1 com um valor de IC50 de 2,9 μM.
Populações especiais
Comprometimento renal
Não foram realizados estudos farmacocinéticos em pacientes com comprometimento renal ou submetidos a diálise. O modelo farmacocinético populacional revelou que a depuração da creatinina não explicou a variabilidade entre os pacientes, pelo que não se recomendam alterações da dose para pacientes com comprometimento renal leve a moderado.
Comprometimento hepático
O neratinibe é extensamente metabolizado no fígado. Em indivíduos com comprometimento hepático preexistente grave (Classe C de Child Pugh) sem câncer, a depuração do neratinibe reduziu em 36% e a exposição ao neratinibe aumentou cerca de 3 vezes, em comparação com voluntários saudáveis.
Dados de segurança pré-clínica
As reações adversas não observadas durante os estudos clínicos, mas constatadas nos animais sujeitos a níveis de exposição análogos aos níveis de exposição clínica, e com eventual relevância para a utilização clínica, foram as seguintes:
Carcinogênese, mutagênese
Maleato de Neratinibe não foi clastogênico nem mutagênico na bateria padrão de testes de genotoxicidade.
Os metabolitos de M3, M6, M7 e M11 do neratinibe são negativos na bateria padrão de testes de genotoxicidade in vitro.
Um estudo de carcinogenicidade de 6 meses em camundongos transgênicos Tg.rasH2 e os dados de 2 anos em ratos não revelaram quaisquer sinais de potencial carcinogênico.
Toxicidade reprodutiva
Em coelhos, não se verificaram efeitos no acasalamento ou na capacidade de os animais engravidarem; no entanto, foram observadas letalidade embriofetal e anomalias morfológicas fetais (por ex., cabeça abaulada, dilatação dos ventrículos cerebrais e fontanelas anteriores deformadas e fontanelas anteriores e/ou posteriores alargadas) em doses que podem ser consideradas clinicamente relevantes.