 Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)
Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)Quais cuidados devo ter ao usar o Mibo?
O Bortezomibe deve ser administrado sob a supervisão de médico com experiência no uso de tratamento antineoplásico.
Ocorreram casos fatais de administração inadvertida de Bortezomibe pela via intratecal. O Bortezomibe deve ser administrado somente pelas vias intravenosa e subcutânea.
O Bortezomibe NÃO DEVE SER ADMINISTRADO PELA VIA INTRATECAL.
Em geral, o perfil de segurança de pacientes tratados com Bortezomibe em monoterapia foi similar ao observado em pacientes tratados com Bortezomibe combinado com melfalana e prednisona.
Neuropatia periférica
O tratamento com Bortezomibe causa neuropatia periférica que é predominantemente sensorial. Entretanto, foram relatados casos de neuropatia motora grave com ou sem neuropatia sensorial periférica.
Pacientes com sintomas pré-existentes (dormência, dor ou sensação de queimação nos pés ou mãos) e/ou sinais de neuropatia periférica podem apresentar piora da neuropatia periférica (incluindo Grau ≥ 3) durante o tratamento com Bortezomibe. Os pacientes devem ser monitorados quanto aos sintomas de neuropatia, como sensação de queimação, hiperestesia, hipoestesia, parestesia, desconforto, dor neuropática ou fraqueza.
No estudo Fase 3 comparando Bortezomibe IV versus SC, a incidência de eventos de neuropatia periférica Grau ≥ 2 foi de 24% para SC e de 41% para IV (p = 0,0124). Neuropatia periférica Grau ≥ 3 ocorreu em 6% dos pacientes no grupo de tratamento SC, comparado a 16% no grupo de tratamento IV (p = 0,0264) (Tabela 17). Portanto, pacientes com neuropatia periférica pré-existente ou com alto risco de neuropatia periférica podem se beneficiar com o uso de Bortezomibe subcutâneo.
Pacientes que apresentarem piora ou aparecimento de neuropatia periférica podem exigir uma mudança de dose, esquema de tratamento ou via de administração para SC. Após o ajuste de doses, a melhora ou resolução da neuropatia periférica foi relatada em 51% dos pacientes com neuropatia periférica Grau ≥ 2 no estudo Fase 3 com agente único, de Bortezomibe versus dexametasona.
A melhora ou resolução da neuropatia periférica foi relatada em 73% dos pacientes que descontinuaram o medicamento devido à neuropatia periférica Grau 2 ou que apresentaram neuropatia periférica Grau ≥ 3 nos estudos Fase 2.
Hipotensão
Em estudos Fase 2 e 3 como agente único para o tratamento de mieloma múltiplo, a incidência de hipotensão (postural, ortostática e hipotensão inespecífica) foi de 11 a 12%. Estes eventos são observados ao longo do tratamento.
Recomenda-se cautela ao tratar pacientes com história de síncope, pacientes recebendo medicamentos sabidamente associados com hipotensão e pacientes desidratados. A conduta na hipotensão ortostática/postural deve incluir ajuste da medicação anti-hipertensiva, hidratação ou administração de mineralocorticoides e/ou simpatomiméticos.
Alterações cardíacas
Desenvolvimento agudo ou exacerbação de insuficiência cardíaca congestiva e/ou início de redução da fração de ejeção do ventrículo esquerdo têm sido relatados, incluindo relatos em pacientes com pouco ou nenhum risco de redução da fração de ejeção do ventrículo esquerdo. Pacientes com fatores de risco ou com doença cardíaca pré-existente devem ser cuidadosamente monitorados.
Em um estudo Fase 3 com agente único, de Bortezomibe versus dexametasona, a incidência de qualquer alteração cardíaca que aparece com o tratamento foi de 15% e 13%, respectivamente. A incidência de eventos de insuficiência cardíaca (edema pulmonar agudo, insuficiência cardíaca, insuficiência cardíaca congestiva, choque cardiogênico e edema pulmonar) foi similar nos grupos de Bortezomibe e dexametasona, 5% e 4%, respectivamente. Houve casos isolados de prolongamento do intervalo QT em estudos clínicos; a causalidade não foi estabelecida.
Eventos hepáticos
Têm sido relatados casos raros de insuficiência hepática aguda em pacientes recebendo medicações concomitantes múltiplas e com sérias condições médicas de base. Outros eventos adversos relatados incluem aumento das enzimas hepáticas, hiperbilirrubinemia e hepatite. Estas alterações podem ser reversíveis com a descontinuação de Bortezomibe. Há informações limitadas relacionadas à reexposição nestes pacientes.
Distúrbios pulmonares
Houve casos raros relatados de doença pulmonar infiltrante difusa aguda de etiologia desconhecida, tais como pneumonite, pneumonia intersticial, infiltração pulmonar, Síndrome do Desconforto Respiratório Agudo (ARDS) em pacientes recebendo Bortezomibe.
Alguns desses eventos têm sido fatais. Uma proporção mais elevada desses efeitos foi relatada no Japão. Na ocorrência de um evento pulmonar ou na piora de sintomas pulmonares já existentes, uma rápida avaliação diagnóstica deve ser realizada e os pacientes tratados apropriadamente.
Em um estudo clínico, dois pacientes que receberam altas doses de citarabina (2g/m2 por dia) por infusão contínua com daunorrubicina e Bortezomibe para recaída de leucemia mieloide aguda morreram com ARDS precocemente durante o tratamento.
Exames laboratoriais
O resultado do hemograma completo deve ser frequentemente monitorado durante o tratamento com Bortezomibe.
Trombocitopenia / Neutropenia
O Bortezomibe está associado com trombocitopenia e neutropenia. As plaquetas tiveram seu nível mais baixo no Dia 11 de cada ciclo de tratamento com Bortezomibe e normalmente recuperaram seu nível basal no próximo ciclo. O padrão cíclico de redução e recuperação da contagem de plaquetas permanece consistente nos estudos de mieloma múltiplo, com nenhuma evidência de trombocitopenia ou neutropenia cumulativas em nenhum dos regimes estudados.
A contagem de plaquetas deve ser monitorada antes de cada dose de Bortezomibe. O tratamento deve ser interrompido quando a contagem de plaquetas for < 25.000/mcL. Existem relatos de hemorragia gastrintestinal e intracerebral associadas com Bortezomibe. Transfusão e cuidados de suporte devem ser considerados.
No estudo de mieloma múltiplo com Bortezomibe como agente único versus dexametasona, a média das contagens mais baixas de plaquetas foi aproximadamente 40% da condição basal. A gravidade da trombocitopenia relacionada à contagem de plaquetas antes do tratamento está na Tabela 10 para estudos de Fase 3 com agente único. A incidência de eventos de sangramento significativo (≥ Grau 3) foi similar em ambos os braços Bortezomibe (4%) e dexametasona (5%).
Tabela 11: Gravidade da trombocitopenia relacionada à contagem de plaquetas antes do tratamento nos estudos Fase 3 com agente único de Bortezomibe vs dexametasona
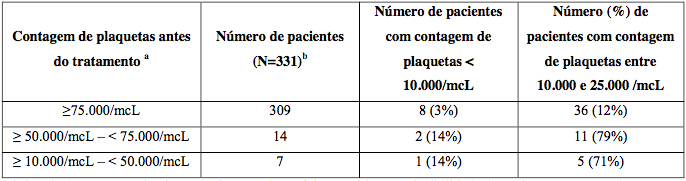
a Níveis basais de contagem de plaquetas de 50000/mcL foi requerida para elegibilidade do paciente para o estudo.
b Faltam dados de contagem basal para 1 paciente.
Eventos adversos gastrintestinais
O tratamento com Bortezomibe pode causar náusea, diarreia, constipação e vômito que exigem, algumas vezes, uso de anti-heméticos e medicamentos antidiarreicos. A reposição de líquidos e eletrólitos deve ser realizada para evitar a desidratação.
Uma vez que alguns pacientes em tratamento com Bortezomibe podem apresentar vômito e/ou diarreia, os pacientes devem ser orientados como proceder para evitar a desidratação. Os pacientes devem ser instruídos para procurar o médico se apresentarem sintomas de vertigem, tontura ou desmaios.
Síndrome da lise tumoral
Uma vez que Bortezomibe é um agente citotóxico e pode matar células malignas rapidamente, as complicações da síndrome da lise tumoral podem ocorrer. Os pacientes sob risco de síndrome da lise tumoral são aqueles com carga tumoral alta antes do tratamento. Estes pacientes devem ser monitorados de perto e as precauções apropriadas devem ser tomadas.
Pacientes com insuficiência hepática
O Bortezomibe é metabolizado pelas enzimas hepáticas e sua exposição é aumentada em pacientes com insuficiência hepática moderada ou grave. Esses pacientes devem ser tratados com doses iniciais reduzidas de Bortezomibe e monitorados com relação à toxicidade.
Síndrome de encefalopatia posterior reversível (SEPR)
Foram relatados casos de síndrome de encefalopatia posterior reversível (SEPR) em pacientes recebendo Bortezomibe. SEPR é um distúrbio neurológico raro, reversível, que pode se apresentar com convulsões, hipertensão, cefaleia, letargia, confusão mental, cegueira, entre outros distúrbios visuais e neurológicos. Exames de imagem do cérebro, preferencialmente RMN (Ressonância Magnética Nuclear) são usados para confirmar o diagnóstico.
Em pacientes com SEPR em desenvolvimento, descontinue Bortezomibe. A segurança em reiniciar o tratamento com Bortezomibe em pacientes com histórico de SEPR não é conhecida.
Carcinogênese, mutagênese, comprometimento da fertilidade
Não foram conduzidos estudos de carcinogenicidade com Bortezomibe.
O Bortezomibe demonstrou atividade clastogênica (aberrações cromossômicas estruturais) em teste in vitro de aberrações cromossômicas usando células de ovário de hamster Chinês. O Bortezomibe não foi genotóxico no teste in vitro de mutagenicidade (teste de Ames) e no teste in vivo de micronúcleos em camundongos.
Não foram realizados estudos de fertilidade com Bortezomibe, mas foi realizada avaliação dos tecidos reprodutivos nos estudos de toxicidade geral. No estudo de toxicidade de 6 meses em rato, foram observados efeitos degenerativos no ovário em doses ≥ 0,3 mg/m2 (um quarto da dose clínica recomendada) e alterações degenerativas nos testículos ocorreram com 1,2 mg/m2. O Bortezomibe pode ter um potencial efeito sobre a fertilidade masculina ou feminina.
Achados de toxicidade em animais
Toxicidade cardiovascular:
Estudos em macacos mostraram que a administração de doses aproximadamente o dobro da dose clínica recomendada resultaram em aumento da frequência cardíaca seguido de significante hipotensão progressiva, bradicardia e morte 12-14 horas após a administração. Doses ≥ 1,2 mg/m2 induziram alterações proporcionais à dose nos parâmetros cardíacos. O Bortezomibe distribuiu-se para a maioria dos tecidos, incluindo o miocárdio. Em um estudo de toxicidade de dose repetida em macaco também foram observadas hemorragia, inflamação e necrose miocárdica.
Administração crônica:
Em estudos em animais em dose e esquema posológico similar ao recomendado para pacientes (duas vezes por semana, durante duas semanas, seguido de uma semana de descanso), os sinais de toxicidade observados incluíram anemia grave e trombocitopenia, toxicidade gastrintestinal, neurológica e do sistema linfático. Efeitos neurotóxicos em estudos animais incluíram edema axonal e degeneração em nervos periféricos, raízes espinhais dorsais e tratos da medula espinhal. Adicionalmente, hemorragia multifocal e necrose no cérebro, olho e coração foram observadas.
População especial
Gravidez (Categoria D)
Mulheres em idade fértil devem evitar a gravidez durante o tratamento com Bortezomibe.
O Bortezomibe não foi teratogênico em estudos pré-clínicos de toxicidade sobre o desenvolvimento em ratos e coelhos na maior dose testada [0,075 mg/kg (0,5 mg/m2) em ratos e 0,05 mg/kg (0,6 mg/m2) em coelhos] quando administrado durante a organogênese. Estas doses são aproximadamente a metade da dose clínica de 1,3 mg/m2 com base na área de superfície corporal.
Coelhas prenhas que receberam 0,05 mg/kg (0,6 mg/m2) de Bortezomibe durante a organogênese apresentaram perda pós-implantação significante e número reduzido de fetos vivos. Os fetos vivos destas ninhadas também apresentaram reduções significantes no peso fetal. A dose é aproximadamente metade da dose clínica de 1,3 mg/m2 com base na área de superfície corporal.
Não foram conduzidos estudos de transferência placentária de Bortezomibe. Não existem estudos controlados em mulheres grávidas. Se Bortezomibe for utilizado durante a gestação ou se a paciente engravidar durante o tratamento, ela deve ser informada sobre o potencial risco para o feto.
As pacientes devem ser orientadas sobre o uso de medidas contraceptivas eficazes e para evitar a amamentação durante o tratamento com Bortezomibe.
Este medicamento não deve ser utilizado por mulheres grávidas sem orientação médica. O médico deverá ser informado imediatamente em caso de suspeita de gravidez.
Lactação
Não existem dados sobre a excreção de Bortezomibe no leite humano. Uma vez que muitos fármacos são excretados no leite humano e devido ao potencial para reações adversas graves em lactentes devido ao Bortezomibe, as mulheres devem ser alertadas para não amamentar durante o tratamento com Bortezomibe.
Efeitos sobre a capacidade de dirigir veículos e operar máquinas
Uma vez que o Bortezomibe pode estar associado à fadiga, tontura, síncope, hipotensão ortostática/postural, diplopia ou visão turva, os pacientes devem ser orientados para não dirigir veículos ou operar máquinas se houver ocorrência de qualquer destes sintomas.
Uso em idosos, crianças e outros grupos de risco
Não foram observadas diferenças gerais em segurança e efetividade entre pacientes com idade ≥ 65 anos e pacientes mais novos recebendo Bortezomibe; entretanto, uma maior sensibilidade de alguns pacientes mais velhos não pode ser afastada. A segurança e a eficácia de Bortezomibe em crianças não foram estabelecidas para mieloma múltiplo.