 Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)
Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)Qual a ação da substância do Mektovi?
Resultados de Eficácia
Eficácia e segurança clínica
Melanoma irressecável ou metastático com mutação BRAF V600
A segurança e a eficácia do binimetinibe em combinação com o encorafenibe foram avaliadas em um estudo Fase III, multicêntrico, randomizado (1:1:1), ativo-controlado, aberto de duas partes, em pacientes com melanoma irressecável ou metastático com mutação BRAF V600E ou K (Estudo CMEK162B2301), detectada usando um teste para mutação BRAF. Os pacientes apresentaram melanoma primário cutâneo ou desconhecido confirmado histologicamente, porém aqueles que apresentavam melanoma uveal ou mucoso foram excluídos.
Os pacientes foram autorizados a receber terapia prévia adjuvante e uma linha prévia de imunoterapia para doença irressecável localmente avançada ou metastática. O tratamento prévio com inibidores de BRAF/MEK não foi permitido.
Estudo CMEK162B2301, parte 1
Na parte 1, os pacientes do Estudo foram randomizados para receber binimetinibe 45 mg via oral duas vezes ao dia e encorafenibe 450 mg via oral diariamente (Combo 450, n=192), encorafenibe 300 mg via oral diariamente (Enco 300, n=194), ou vemurafenibe 960 mg via oral duas vezes ao dia (daqui em diante referido como Vem, n=191). O tratamento continuou até a progressão da doença ou toxicidade inaceitável. A randomização foi estratificada pela classificação do Comitê Americano de Câncer (AJCC - American Joint Committee on Cancer) (IIIB, IIIC, IVM1a ou IVM1b, vs IVM1c) e escala de desempenho ECOG - Eastern Cooperative Oncology Group (0 versus 1) e imunoterapia prévia para doença irressecável ou metastática (sim versus não).
O desfecho primário de eficácia foi a sobrevida livre de progressão (SLP) do Combo 450 em comparação com o vemurafenibe, conforme avaliado por um comitê de revisão independente cego (BIRC). A SLP avaliada pelos investigadores (avaliação do investigador) foi uma análise de apoio. Um desfecho secundário adicional incluiu a SLP do Combo 450 em comparação com o Enco 300. Outras comparações secundárias de eficácia entre o Combo 450 e o vemurafenibe ou o Enco 300 incluíram sobrevida global (SG), taxa de resposta objetiva (TRO), duração da resposta (DDR) e taxa de controle de doença (TCD) avaliadas pelo BIRC e pela avaliação do investigador.
A idade mediana dos pacientes foi de 56 anos (variação de 20-89), 58% eram do sexo masculino, 90% eram caucasianos e 72% tinham PS-ECOG 0 no basal. A maioria dos pacientes tinha doença metastática (95%) e estágio IVM1c (64%); 27% apresentavam desidrogenase láctica (DHL) elevada, e 45% tinham pelo menos 3 órgãos envolvidos no basal e 3,5% tinham metástases cerebrais. Vinte e sete pacientes (5%) receberam previamente inibidores de ponto de verificação (anti-PD1/PDL1 ou ipilimumabe) (8 pacientes no grupo Combo 450 (4%), 7 no braço do vemurafenibe (4%) e 12 no braço do Enco (braço do Combo 450, 5 pacientes no braço do vemurafenibe, 11 no Enco 300) e 5 pacientes no esquema adjuvante (2 no braço do Combo 450, 2 no vemurafenibe, 1 no Enco 300).
A duração mediana da exposição foi de 11,7 meses em pacientes tratados com Combo 450, 7,1 meses no Enco 300 e 6,2 meses no vemurafenibe. A mediana da intensidade relativa da dose para o Combo 450 foi de 100% para o encorafenibe e 99,6% para o binimetinibe, 86,2% para o Enco 300 e 94,5% para o vemurafenibe.
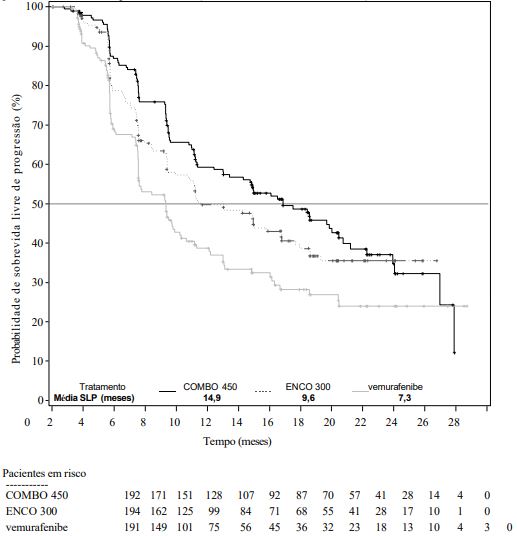
A parte 1 do Estudo CMEK162B2301 demonstrou melhora estatisticamente significativa na SLP nos pacientes tratados com Combo 450 em comparação com os pacientes tratados com vemurafenibe. A Tabela 1 e a Figura 1 resumem a SLP e outros resultados de eficácia baseados na revisão central dos dados por um comitê cego independente de radiologia.
Os resultados de eficácia baseados na avaliação do investigador foram consistentes com a avaliação central independente (BIRC). Análises não estratificadas de subgrupos demonstraram pontos de estimativa a favor do Combo 450, incluindo DHL basal, escala de desempenho ECOG e estágio de AJCC.
Tabela 1: Estudo CMEK162B2301, parte 1: sobrevida livre de progressão e resultados confirmados de resposta geral pelo BIRC (revisão central independente)
| Encorafenibe + binimetinibe N=192 (Combo 450) | Encorafenibe N=194 (Enco 300) | Vemurafenibe N=191 (Vem) | |
| Data de corte: 19 de maio de 2016 | |||
| SLP (análise primária) | |||
| Número de eventos [doença progressiva (DP)] (%) | 98 (51,0) | 96 (49,5) | 106 (55,5) |
| Mediana, meses | 14,9 | 9,6 | 7,3 |
| (IC 95%) | (11,0, 18,5) | (7,5, 14,8) | (5,6, 8,2) |
| Razão de Risco (Hazard ratio)a (IC 95%) (vs Vem) | 0,54 (0,41, 0,71) | ||
| valor-p (log-rank estratificado)b | <0,001 | ||
| Razão de Risco (Hazard ratio)a (IC 95%) (vs Vem) | 0,68 (0,52, 0,90) | ||
| valor-p nominal | 0,007 | ||
| Razão de Risco (Hazard ratio)a (IC 95%) (vs Enco 300) | 0,75 (0,56, 1,00) | ||
| valor-p (log-rank estratificado)b | 0,051 | ||
| Respostas gerais confirmadas | |||
| Taxa de respostas gerais, n (%) | 121 (63,0) | 98 (50,5) | 77 (40,3) |
| (IC 95%) | (55,8, 69,9) | (43,3, 57,8) | (33,3, 47,6) |
| RC, n (%) | 15 (7,8) | 10 (5,2) | 11 (5,8) |
| RP, n (%) | 106 (55,2) | 88 (45,4) | 66 (34,6) |
| DE, n (%) | 46 (24,0) | 53 (27,3) | 73 (38,2) |
| TCD, n (%) | 177 (92,2) | 163 (84,0) | 156 (81,7) |
| (IC 95%) | (87,4, 95,6) | (78,1, 88,9) | (75,4, 86,9) |
| Duração da resposta | |||
| Mediana, meses | 16,6 | 14,9 | 12,3 |
| (IC 95%) | (12,2, 20,4) | (11,1, NE) | (6,9, 16,9) |
| Análise atualizada, data de corte: 07 de novembro de 2017 | |||
| SLP | |||
| Número de eventos (doença progressiva) (%) | 113 (58,9) | 112 (57,7) | 118 (61,8) |
| Mediana, meses | 14,9 | 9,6 | 7,3 |
| (IC 95%) | (11,0, 20,2) | (7,4, 14,8) | (5,6, 7,9) |
| Razão de Risco (Hazard ratio)a (IC 95%) (vs Vem) | 0,51 (0,39, 0,67) | ||
| valor-p nominal | <0,001 | ||
| Razão de Risco (Hazard ratio)a (IC 95%) (vs Vem) | 0,68 (0,52, 0,88) | ||
| valor-p nominal | 0,0038 | ||
| Razão de Risco (Hazard ratio)a (IC 95%) (vs Enco300) | 0,77 (0,59, 1,00) | ||
| valor-p nominal | 0,0498 | ||
IC= intervalo de confiança; RC= resposta completa; TCD= taxa de controle de doença (RC+RP+DE+nãoRC/não-DP; não-RC/não-DP aplica-se apenas a pacientes sem uma lesão alvo que não atingiu RC ou tem DP); HR= razão de risco; NE= não estimável; SLP: sobrevida livre de progressão; RP= resposta parcial; DE= doença estável. Vem= vemurafenibe.
a Razão de risco baseado em um modelo de risco proporcional estratificado de Cox.
b Valor-p log-rank (2 lados).
Figura 1. Estudo CMEK162B2301, parte 1: Gráfico de Kaplan-Meier de sobrevida livre de progressão por revisão central independente BIRC (data de corte: 19 de maio de 2016)
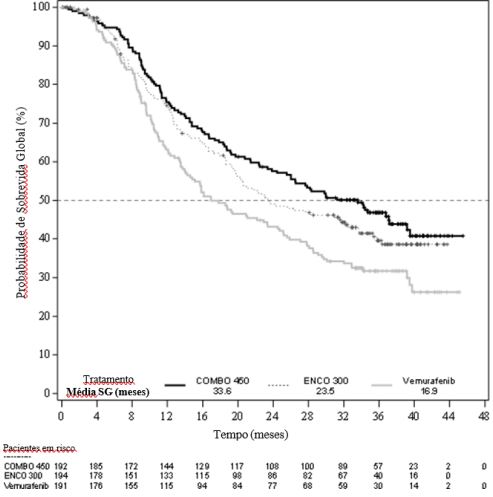
A análise preliminar da SG do Estudo CMEK162B2301 parte 1, (data de corte 07 de novembro de 2017) demonstrou melhora estatisticamente significativa na SG para o Combo 450 em comparação com o vemurafenibe (vide Tabela 2 e Figura 2).
Uma proporção semelhante de pacientes em cada grupo recebeu tratamento subsequente com inibidores de ponto de verificação, principalmente pembrolizumabe, nivolumabe e ipilimumabe (34,4% do braço do Combo 450, 36,1% do braço do encorafenibe, 39,8% do vemurafenibe).
Tabela 2: Estudo CMEK162B2301, parte 1: Resultados preliminares de sobrevida global (data de corte: 07 de novembro de 2017)
| Encorafenibe + binimetinibe N=192 (Combo 450) | Encorafenibe N=194 (Enco 300) | Vemurafenibe N=191 (Vem) | |
| SG | |||
| Número de eventos (%) | 105 (54,7) | 106 (54,6) | 127 (66,5) |
| Mediana, meses | 33,6 | 23,5 | 16,9 |
| (IC 95%) | (24,4, 39,2) | (19,6, 33,6) | (14,0, 24,5) |
| Sobrevida em 12 meses | 75,5% | 74,6% | 63,1% |
| (IC 95%) | (68,8, 81,0) | (67,6, 80,3) | (55,7, 69,6) |
| Sobrevida em 24 meses | 57,6% | 49,1% | 43,2% |
| (IC 95%) | (50,3, 64,3) | (41,5, 56,2) | (35,9, 50,2) |
| Razão de Risco (Hazard ratio)a (IC 95%) (vs Vem) | 0,61 (0,47, 0,79) | ||
| valor-p (log-rank estratificado) | <0,0001 | ||
| Razão de Risco (Hazard ratio)a (IC 95%) (vs Enco 300) | 0,81 (0,61, 1,06) | ||
| valor-p (log-rank estratificado) | 0,061 | ||
Figura 2: Estudo CMEK162B2301, parte 1: Gráfico de Kaplan-Meier de resultados preliminares de sobrevida global (data de corte: 07 de novembro de 2017)
Qualidade de Vida (QdV) (data de corte: 19 de maio de 2016)
Para explorar as medidas de Qualidade Vida relacionada à saúde, funcionalidade, sintomas do melanoma e reações adversas relacionadas ao tratamento dos resultados relatados pelos pacientes (RRP), foram utilizados a Avaliação Funcional para Terapia de Câncer - Melanoma (FACT-M - Functional Assessment of Cancer Therapy Melanoma), o Questionário de Qualidade de Vida da Organização Européia para Pesquisa e Tratamento do Câncer (EORTC QLQ-C30 - European Organisation for Research and Treatment of Cancer's core quality of life questionnaire) e o exame EuroQoL-5dimensões-5níveis (EQ-5D-5L). Uma deterioração definitiva de 10% no FACT-M e no EORTC QLQ-C30 foi significativamente retardado nos pacientes tratados com Combo 450 em relação a outros tratamentos. A mediana do tempo para a deterioração definitiva de 10% na escala FACT-M não foi alcançada no braço do Combo 450 e foi de 22,1 meses (IC 95%: 15.2, NE) no braço do vemurafenibe com um HR para a diferença de 0.46 (IC 95%: 0.29, 0.72). Uma análise do tempo de deterioração definitiva de 10% na escala EORTC QLQ-C30 forneceu resultados semelhantes.
Os pacientes que receberam o Combo 450 não relataram mudança ou leve melhora na variação média em relação à pontuação inicial da escala EQ-5D-5L em todas as consultas, enquanto os que receberam vemurafenibe ou encorafenibe referiram piora em todas as visitas (com diferenças estatisticamente significativas). Uma avaliação de mudança ao longo do tempo na pontuação rendeu a mesma tendência para o EORTC QLQ-C30 e em todas as visitas para o FACT-M.
Estudo CMEK162B2301, parte 2
A parte 2 do Estudo CMEK162B2301 foi projetada para avaliar a contribuição do binimetinibe para a combinação do encorafenibe e binimetinibe.
A SLP para o encorafenibe 300 mg via oral diariamente utilizada em combinação com o binimetinibe 45 mg via oral duas vezes ao dia (Combo 300, n=258) foi comparada com a SLP para o Enco 300 (n=280, incluindo 194 pacientes da parte 1 e 86 pacientes da parte 2). A inclusão na parte 2 iniciou após todos os pacientes da parte 1 serem randomizados.
Dados preliminares da parte 2, na data de corte de 09 de novembro de 2016, demonstraram a contribuição do binimetinibe com uma estimativa de mediana de SLP de 12,9 meses (IC 95%: 10.1, 14.0) para o Combo 300 em comparação com 9,2 meses (IC 95%: 7.4, 11.0) para o Enco 300 (partes 1 e 2) pela revisão do comitê central independente (BIRC). Resultados similares foram observados pela avaliação do investigador.
A TRO confirmada por BIRC foi de 65,9% (IC 95%: 59.8, 71.1) para o Combo 300 e 50,4% (IC 95%: 44.3, 56.4) para o Enco 300 (partes 1 e 2). A mediana da DDR para respostas confirmadas pelo BIRC foi de 12,7 meses [IC 95%: 9.3, 15.1] para o Combo 300 e 12,9 meses [IC 95%: 8.9, 15.5] para o Enco 300. A mediana da duração do tratamento foi maior para o Combo 300 versus Enco 300, 52,1 semanas versus 31,5 semanas.
Eletrofisiologia Cardíaca
Na análise de segurança de estudos agrupados de encorafenibe 450 mg uma vez ao dia em combinação com 45 mg de binimetinibe duas vezes ao dia (Combo 450), a incidência de novo prolongamento de QTc>500 ms foi de 0,7% (2/268), enquanto no grupo encorafenibe com agente único foi de 2,5% (5/203). O prolongamento de Qtc>60ms comparado aos valores do pré-tratamento foi observado em 4,9% (13/268) dos pacientes no grupo encorafenibe mais binimetinibe e em 3,4% (7/204) no grupo encorafenibe como agente único.
Referências bibliográficas
1. Drummer R, Ascierto P, Gojas H, et al. Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): a multicentre, open-label, randomised phase 3 trial. Lancet http://dx.doi.org/10.1016/S1470-2045(18)30142-6.
2. Drummer R, Ascierto P, Gojas H, et al. Overall survival in patients with BRAF-mutant melanoma receiving encorafenib plus binimetinib versus vemurafenib or encorafenib (COLUMBUS): a multicentre, open-label, randomised, phase 3 trial. Lancet http://dx.doi.org/10.1016/S1470-2045(18)30497-2.
Características Farmacológicas
Propriedades farmacodinâmicas
Grupo farmacoterapêutico: agente antineoplásico, inibidor de proteína quinase, código ATC: L01XE41.
Mecanismo de ação
O binimetinibe é um inibidor reversível de atividade de quinase, não competitivo de ATP, da quinase regulada por sinal extracelular ativada por mitógeno 1 (MEK1) e MEK2. No sistema isento de célula, o binimetinibe inibe a MEK1 e MEK2 com a metade da concentração inibitória máxima (IC50) em 12-46 nM. As proteínas MEK são reguladoras acima da via da quinase relacionada ao sinal extracelular (ERK), que promove a proliferação celular. No melanoma e em outros cânceres, esta via é frequentemente ativada por formas mutadas de BRAF que ativam a MEK. O binimetinibe inibe a ativação da MEK pelo BRAF e inibe a atividade da MEK quinase. O binimetinibe inibe o crescimento de linhas celulares de melanoma com mutação BRAF V600 e demonstra efeitos antitumorais em modelos animais do melanoma com mutação BRAF V600.
Combinação com encorafenibe
O binimetinibe e o encorafenibe (um inibidor de BRAF, vide Características Farmacológicas da bula encorafenibe) inibem a via MAPK, resultando em uma atividade antitumoral maior.
Adicionalmente, a combinação entre encorafenibe e binimetinibe preveniu o aparecimento de resistência in vivo nos xenoenxertos de melanoma humano com mutação BRAF V600E.
Propriedades farmacocinéticas
A farmacocinética do binimetinibe foi estudada em indivíduos saudáveis e em pacientes com tumores sólidos e melanoma cutâneo avançado e irressecável ou metastático. Após repetir a administração duas vezes ao dia concomitante com o encorafenibe, as condições de estado estacionário do binimetinibe foram alcançadas em 15 dias, sem acúmulo importante. A média (CV%) de Cmax ss foi de 654 ng/mL (34,7%) e a média ASCss foi de 2,35 μg/mL (28,0%) em combinação com o encorafenibe, conforme estimado pela modelagem PK da população. A farmacocinética do binimetinibe demonstrou ser aproximadamente linear na dose.
Absorção
Após a administração oral, o binimetinibe é rapidamente absorvido com uma mediana de Tmax de 1,5 horas. Após uma dose oral única de 45 mg [14C] de binimetinibe em indivíduos saudáveis, no mínimo 50% da dose foi absorvida. A administração de uma dose única de 45 mg de binimetinibe com refeições altamente calóricas e com alto índice de gordura diminuíram a concentração máxima de binimetinibe (Cmax) para 17%, enquanto a área sob a curva (ASC) de tempo de concentração permaneceu inalterada. O Estudo de interação com o medicamento em indivíduos saudáveis indica que a exposição de binimetinibe não é alterada na presença de um agente que altera o pH gástrico (rabeprazol).
Distribuição
O binimetinibe se liga a 97,2% das proteínas plasmáticas humanas in vitro. O binimetinibe é mais distribuído no plasma do que no sangue. Em humanos, a relação plasma-sangue é de 0,718. Após uma dose oral única de 45 mg [14C] de binimetinibe em indivíduos saudáveis, o volume aparente de distribuição (Vz/F) do binimetinibe é de 374 L.
Biotransformação
Após uma dose oral única de 45 mg [14C] de binimetinibe em indivíduos saudáveis, as vias de biotransformação primária do binimetinibe observadas em humanos incluem glucuronidação, N-desalquilação, hidrólise amida e perda do etanodiol da cadeia lateral. A contribuição máxima da glucuronidação direta para o clearence de binimetinibe foi estimada em 61,2%. Após uma dose oral única de 45 mg [14C] de binimetinibe em indivíduos saudáveis, aproximadamente 60% da ASC da radioatividade circulante no plasma foi atribuída ao binimetinibe. In vitro, a CYP1A2 e CYP2C19 catalisam a formação do metabólito ativo, o qual representa menos de 20% da exposição do binimetinibe clinicamente.
Eliminação
Após uma dose oral única de 45 mg [14C] de binimetinibe em indivíduos saudáveis, uma média de 62,3% da radioatividade foi eliminada nas fezes enquanto 31,4% foi eliminada na urina. Nesta última, 6,5% da radioatividade foi excretada como binimetinibe. A média (CV%) de clearance aparente (CL/F) do binimetinibe foi de 28,2 L/h (17,5%). A mediana (intervalo) da meia-vida terminal do binimetinibe (T1/2) foi de 8,66h (8,10 a 13,6h).
Interações medicamentosas
Efeitos dos indutores ou inibidores de UGT1A1 sobre o binimetinibe
O binimetinibe é metabolizado principalmente através da glucuronidação mediada por UGT1A1. Na subanálise do estudo clínico, no entanto, não foi observada uma relação aparente entre a exposição ao binimetinibe e o estado de mutação do UGT1A1. Além disso, simulações para investigar o efeito de 400 mg de atazanavir (inibidor de UGT1A1) na exposição de 45 mg de binimetinibe previram Cmax de binimetinibe semelhante na presença ou ausência de atazanavir. Portanto, a extensão das interações medicamentosas mediadas pelo UGT1A1 é mínima e improvável clinicamente relevante; entretanto, como isso não foi avaliado em um estudo clínico formal, os indutores ou inibidores de UGT1A1 devem ser administrados com cautela.
Efeito de enzimas CYP sobre o binimetinibe
In vitro, a CYP1A2 e a CYP2C19 catalisam a formação do metabólito ativo AR00426032 (M3) através da Ndesmetilação oxidativa.
Efeito do binimetinibe sobre os substratos da CYP
O binimetinibe é um inibidor reversível fraco de CYP1A2 e CYPP2C9.
Efeito dos transportadores sobre o binimetinibe
Experiências in vitro indicam que o binimetinibe é um substrato da glicoproteína P (P-gp) e da proteína de resistência do tumor de mama (BCRP). É improvável que a inibição da P-gp ou da BCRP resulte em um aumento clinicamente importante nas concentrações de binimetinibe, uma vez que este exibe permeabilidade passiva moderada a alta.
Efeito do binimetinibe sobre os transportadores
O binimetinibe é um inibidor fraco de OAT3. Não é esperada qualquer interação medicamentosa clinicamente significativa causada pelo binimetinibe sobre outros transportadores.
O binimetinibe é metabolizado pelas UGTs e CYP1A2 e é um substrato da P-gp. Indutores específicos dessas enzimas não foram estudados e podem resultar em perda de eficácia.
Populações especiais
População pediátrica
A segurança e eficácia do binimetinibe ainda não foram estabelecidas em crianças e adolescentes. Não há dados disponíveis.
Idade, peso corporal
Baseado em uma análise farmacocinética populacional, a idade ou peso corporal não têm um efeito clinicamente importante na exposição sistêmica do binimetinibe.
Gênero
Com base em uma análise farmacocinética populacional, a farmacocinética de binimetinibe foi semelhante em homens quando comparado com mulheres.
Raça
Não existem dados suficientes para avaliar potenciais diferenças na exposição do binimetinibe por raça ou etnia.
Insuficiência hepática
Como o binimetinibe é principalmente metabolizado e eliminado por via hepática, os pacientes com insuficiência hepática moderada a grave podem ter uma exposição aumentada. Os resultados de um estudo clínico dedicado com binimetinibe indicam apenas exposições semelhantes em pacientes com insuficiência leve (Child-Pugh classe A) e indivíduos com função hepática normal. Observou-se um aumento de duas vezes na exposição total ao binimetinibe (ASC) em pacientes com insuficiência hepática moderada (Child-Pugh classe B) e grave (Child-Pugh classe C). Este aumento vai para três vezes, tanto na insuficiência hepática moderada como na grave, quando se considera a exposição ao binimetinibe não ligado.
Síndrome de Gilbert
O binimetinibe não foi avaliado em pacientes com doença de Gilbert. A principal via de transformação hepática do binimetinibe é a glucuronidação, a decisão pelo o tratamento deve ser tomada pelo médico levando em conta o risco benefício individual.
Insuficiência renal
O binimetinibe sofre mínima eliminação renal. Os resultados de um estudo clínico dedicado mostraram que pacientes com insuficiência renal grave (eGFR ≤ 29 mL/min/1,73 m2 ) tiveram um aumento de 29% na exposição (ASCinf), um aumento de 21% na Cmax e uma diminuição de 22% na CL/F em comparação com a correspondência de indivíduos saudáveis. Estas diferenças estiveram dentro da variabilidade observada para estes parâmetros em ambas as coortes deste estudo (25% - 49%) e da variabilidade anteriormente observada em estudos clínicos de pacientes, sendo que estas diferenças não são clinicamente relevantes.
Os efeitos da insuficiência renal na farmacocinética do binimetinibe em combinação com o encorafenibe não foram avaliados clinicamente.
Dados de segurança pré-clínica
A administração oral repetida de binimetinibe em ratos por até seis meses foi associada à mineralização de tecido mole, lesões da mucosa gástrica e alterações patológicas reversíveis mínimas a leves em 7 a 12,5 vezes as exposições terapêuticas em humanos. Em um estudo de irritação gástrica em ratos, observou-se um aumento na incidência de lesões mucosas superficiais e de úlceras hemorrágicas. Em macacos cynomolgus, a administração oral de binimetinibe foi associada à intolerância gastrointestinal, alterações moderadas da patologia clínica, hipercelularidade da medula óssea e achados microscópicos de inflamação gastrointestinal, reversíveis nas doses mais baixas que estavam abaixo das exposições terapêuticas humanas
. O potencial carcinogênico do binimetinibe não foi avaliado. Os estudos de genotoxicidade padrão com binimetinibe foram negativos.
Os potenciais efeitos embrio-fetais do binimetinibe foram avaliados em ratos e coelhos. Em ratos, observou-se ganho de peso corporal gestacional e peso corporal fetal menores e um número reduzido de esternos fetais ossificados. Nenhum efeito foi observado em 14 vezes a exposição terapêutica humana.
Em coelhos, foram observados mortalidade, sinais físicos maternos de toxicidade, menor peso corporal gestacional e aborto. O número de fetos viáveis e os pesos corporais fetais foram reduzidos e a perda pósimplante e as reabsorções aumentaram. Um aumento na incidência de defeitos do septo ventricular fetal e alterações no tronco pulmonar foram observados nas doses mais altas. Nenhum efeito foi observado em 3 vezes a exposição terapêutica humana.
Os estudos de fertilidade não foram realizados com o binimetinibe. Em estudos de toxicidade de dose repetida, não foi levantada qualquer preocupação em termos de fertilidade do exame patológico dos órgãos reprodutivos em ratos e macacos.
O binimetinibe tem potencial fototóxico in vitro.
Um risco mínimo de fotossensibilização foi demonstrado in vivo em uma dose oral, proporcionando uma exposição 3,8 vezes superior à obtida com a dose recomendada em humanos. Estes dados indicam que existe um risco mínimo de fototoxicidade com binimetinibe em doses terapêuticas em pacientes.