 Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)
Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)Quais as reações adversas e os efeitos colaterais do Venomy?
As reações adversas são apresentadas nesta seção. Reações adversas são eventos adversos que foram considerados razoavelmente associados ao uso de acetato de abiraterona, com base na avaliação abrangente das informações de eventos adversos disponíveis. Em casos individuais, uma relação causal com o acetato de abiraterona não pode ser estabelecida com confiança. Portanto, pelo fato de que os estudos clínicos são conduzidos em condições amplamente variadas, as taxas de reações adversas observadas nos estudos clínicos de um medicamento não podem ser diretamente comparadas com as taxas nos estudos clínicos de outros medicamentos e podem não refletir as taxas observadas na prática clínica.
As reações adversas mais comuns observadas com Acetato de Abiraterona são
Edema periférico, hipopotassemia, infecção do trato urinário, aumentos nos níveis de alanina aminotransferase, aumentos nos níveis de aspartato aminotransferase, dispepsia, hematúria, hipertensão e fraturas.
Acetato de Abiraterona pode causar hipertensão, hipopotassemia e retenção hídrica como consequência farmacodinâmica de seu mecanismo de ação.
Em estudos fase 3, os efeitos mineralocorticoides esperados observados mais frequentemente em pacientes tratados com Acetato de Abiraterona em comparação ao placebo foram
Hipopotassemia (18% versus 11%), hipertensão (15% versus 11%) e retenção hídrica - edema periférico (26% versus 20%), respectivamente. Em pacientes tratados com Acetato de Abiraterona, hipopotassemia e hipertensão, ambas de grau 3 e 4, foram observadas em 4% e 2% dos pacientes, respectivamente. Em geral, foi possível controlar os efeitos mineralocorticoides com medicamentos. O uso concomitante de corticosteroide reduz a incidência e a gravidade destas reações adversas.
Em um estudo fase 3 de pacientes com câncer de próstata avançado metastático (estudo 301) que estavam usando agonista LHRH ou que haviam sido submetidos previamente à orquiectomia, no grupo do tratamento ativo,Acetato de Abiraterona foi administrado na dose de 1000 mg/dia em combinação com dose baixa de prednisona ou prednisolona (10 mg/dia); no grupo controle, foi administrado placebo mais dose baixa de prednisona ou prednisolona (10 mg/dia). Os pacientes incluídos eram intolerantes ou haviam falhado a esquemas prévios de quimioterapia, um dos quais contendo docetaxel. A duração média do tratamento com Acetato de Abiraterona foi 8 meses.
As reações adversas com o uso de Acetato de Abiraterona que ocorreram em taxa >1% (de todos os graus) durante o estudo 301 estão na Tabela 5.
Tabela 5: Reações adversas com o uso de Acetato de Abiraterona em > 1% dos pacientes em um estudo fase 3 (Estudo 301)a:
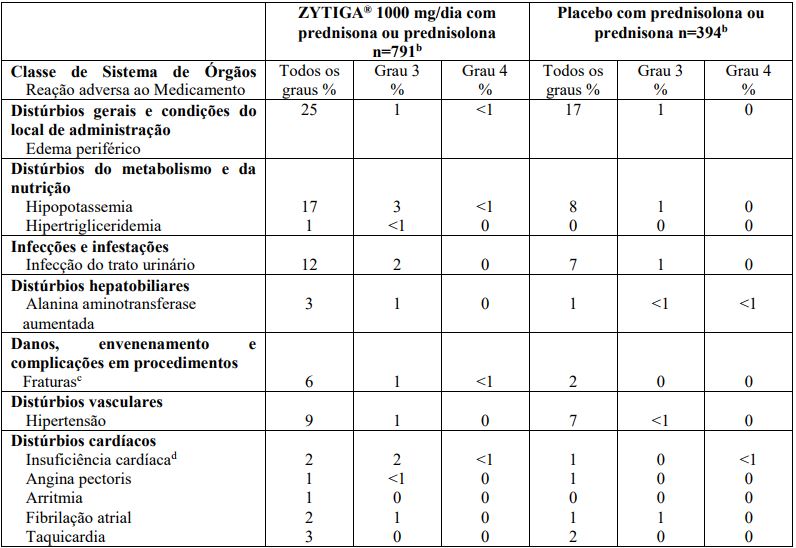 a Todos os pacientes estavam recebendo um agonista de LHRH ou haviam sido submetidos à orquiectomia;
a Todos os pacientes estavam recebendo um agonista de LHRH ou haviam sido submetidos à orquiectomia;
b n= pacientes avaliados para segurança;
c “Fraturas” inclui todas as fraturas, com exceção às fraturas patológicas;
d “Insuficiência cardíaca” também inclui insuficiência cardíaca congestiva, disfunção do ventrículo esquerdo e fração de ejeção diminuída.
Em um segundo estudo clínico fase 3, multicêntrico, controlado com placebo (estudo 302), em pacientes com câncer de próstata avançado metastático assintomático ou levemente sintomático, virgens de quimioterapia, que faziam uso de agonistas de LHRH ou haviam sido submetidos previamente à orquiectomia, Acetato de Abiraterona foi também administrado na dose de 1000 mg ao dia, em associação a uma dose baixa de prednisona ou prednisolona de 10 mg ao dia no braço experimental. Os pacientes do grupo controle receberam placebo e dose baixa de prednisona ou prednisolona de 10 mg ao dia. A duração média do tratamento com Acetato de Abiraterona no estudo 302 foi de 13,8 meses.
As reações adversas com o uso de Acetato de Abiraterona que ocorreram em taxa >1% (de todos os graus) durante o estudo 302 estão na Tabela 6.
Tabela 6: Reações adversas com o uso de Acetato de Abiraterona em > 1% dos pacientes em um estudo fase 3 (Estudo 302)a:
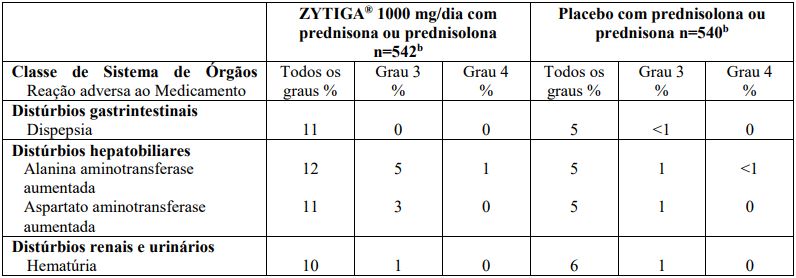 a Todos os pacientes estavam recebendo um agonista de LHRH ou haviam sido submetidos à orquiectomia.
a Todos os pacientes estavam recebendo um agonista de LHRH ou haviam sido submetidos à orquiectomia.
b n= pacientes avaliados para segurança.
As reações adversas mais comuns obtidas dos dados de ambos estudos fase 3 que resultaram na descontinuação do fármaco foram alanina aminotransferase e aspartato aminotransferase aumentadas (cada uma em <1% dos pacientes que receberam Acetato de Abiraterona).
A reação adversa ao medicamento insuficiência da suprarrenal ocorreu nos estudos clínicos fase 3 a uma taxa de 0,5% em pacientes que receberam Acetato de Abiraterona e a uma taxa de 0,2% em pacientes que receberam placebo.
Nos estudos fase 3, 73% dos pacientes tinham idade > 65 anos e 30% tinham idade >75 anos. Não foram observadas diferenças relativas à segurança entre estes pacientes mais idosos e pacientes mais jovens.
Efeitos cardiovasculares
Ambos os estudos fase 3 excluíram pacientes com hipertensão não controlada, doença cardíaca clinicamente significante evidenciada por infarto do miocárdio, eventos de trombose arterial nos últimos 6 meses, angina grave ou instável, insuficiência cardíaca Classe III ou IV da NYHA (estudo 301) ou insuficiência cardíaca Classe II a IV (estudo 302) ou fração de ejeção <50%. Todos os pacientes incluídos (tanto os pacientes tratados com o ativo como com o placebo) foram tratados concomitantemente com tratamento de privação androgênica, predominantemente uso de agonistas de LHRH, a qual foi associada com diabetes, infarto do miocárdio, acidente vascular cerebral e morte súbita.
Nos estudos fase 3, a incidência de reações adversas cardiovasculares em pacientes que receberam Acetato de Abiraterona em comparação aos pacientes que receberam placebo foi a seguinte
Fibrilação atrial (3,4% versus 3,4%); taquicardia (2,8 versus 1,7%); angina pectoris (1,9% versus 0,9%); insuficiência cardíaca (1,9% versus 0,6%) e arritmia (1,1% versus 0,4%).
Hepatotoxicidade
Hepatotoxicidade medicamentosa com níveis elevados de ALT, AST e bilirrubina total foi relatada em pacientes tratados com Acetato de Abiraterona. Entre todos os estudos, elevação dos resultados dos testes de função hepática (aumentos >5 vezes o limite superior da normalidade da ALT ou AST ou aumento >1,5 vezes o limite superior da normalidadeda bilirrubina) foram relatados em aproximadamente 4% dos pacientes que receberam Acetato de Abiraterona, tipicamente durante os 3 primeiros meses após o início do tratamento. No estudo clínico 301, os pacientes com níveis basais elevados de ALT ou AST foram mais propensos a apresentar elevações dos testes de função hepática do que aqueles cujos valores iniciais eram normais. Quando foram observados aumentos da ALT ou AST maior que 5 vezes o limite superior da normalidade ou da bilirrubina maior que 3 vezes o limite superior da normalidade, Acetato de Abiraterona foi suspenso ou descontinuado. Em duas ocasiões ocorreram aumentos acentuados dos testes de função hepática. Estes dois pacientes com função hepática normal na linha de base apresentaram aumentos da ALT ou AST de 15 a 40 vezes o limite superior da normalidade e da bilirrubina de 2 a 6 vezes o limite superior da normalidade. Com a descontinuação de Acetato de Abiraterona, os testes de função hepática normalizaram em ambos os pacientes e um paciente foi tratado novamente com Acetato de Abiraterona, sem recorrência dos aumentos. No estudo clínico 302, elevações de ALT ou AST grau 3 ou 4 foram observadas em 35 (6,5%) dos pacientes tratados com Acetato de Abiraterona. As elevações das aminostransferases se resolveram em todos, exceto em 3 pacientes (2 com novas metástases hepáticas múltiplas e 1 com elevação de AST aproximadamente após 3 semanas da última dose de Acetato de Abiraterona). A descontinuação do tratamento devido à elevação de ALT e AST foi relatada respectivamente em 1,7% e 1,3% dos pacientes tratados com Acetato de Abiraterona e 0,2% e 0% dos pacientes tratados com placebo. Nenhum óbito por hepatotoxicidade foi relatado.
Em estudos clínicos, o risco de hepatotoxicidade foi amenizado pela exclusão de pacientes com hepatite ou anormalidades significativas nos testes de função hepática. No estudo clínico 301, pacientes com ALT e AST basais > 2,5 vezes o limite superior da normalidade, na ausência de metástases hepáticas, e maior que 5 vezes o limite superior da normalidade, na presença de metástases hepáticas, foram excluídos. No estudo clínico 302, pacientes com metástases hepáticas não foram elegíveis e pacientes com ALT e AST basais > 2,5 vezes o limite superior da normalidade foram excluídos. As alterações nos testes de função hepática que se desenvolveram em pacientes participando de estudos clínicos foram vigorosamente manejados pela interrupção do tratamento e permissão para reiniciá-lo apenas depois do retorno dos testes de função hepática ao nível basal do paciente. Os pacientes que apresentaram elevações da ALT ou AST maior que 20 vezes o limite superior da normalidade não reiniciaram o tratamento. A segurança do reinício do tratamento em tais pacientes é desconhecida. O mecanismo para hepatotoxicidade associado com Acetato de Abiraterona não é compreendido.
Experiência de pós-comercialização
As reações adversas apresentadas a seguir, foram identificadas durante a experiência pós-comercialização, com base em relatos espontâneos com o uso de Acetato de Abiraterona.
Classe de Sistema de Órgãos
Distúrbios respiratórios, torácicos e mediastinais
Reação rara (≥ 1/10.000 e < 1/1.000)
Alveolite alérgica.
Distúrbios musculoesqueléticos e do tecido conjuntivo
Reação incomum (≥ 1/1000 and < 1/100)
Rabdomiólise, miopatia.
Distúrbios hepatobiliares
Reação rara (≥ 1/10.000 e < 1/1.000)
Hepatite fulminante, insuficiência hepática aguda.
Em casos de eventos adversos, notifique ao Sistema de Notificações em Vigilância Sanitária - NOTIVISA ou para a Vigilância Sanitária Estadual ou Municipal.