 Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)
Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)Qual a ação da substância do Ritonavir Laboratório Cristália?
Resultados de Eficácia
Comprimido
Danner et al. demonstraram a potência, segurança e eficácia do Ritonavir em um estudo que avaliou regimes contendo diferentes doses de Ritonavir, comparando-os com placebo. Após 32 semanas, o grupo que recebeu 600 mg a cada 12 horas, em combinação com outros agentes ARV (inibidores da transcriptase reversa análogos de nucleosídeo) apresentaram um ganho de CD4 de 230 células por mm3 e uma queda na carga viral de 0,81 log. Os eventos adversos mais comuns foram náuseas, aumento dos triglicérides e das enzimas hepáticas. Assim, o Ritonavir mostrou-se seguro, eficaz e bem tolerado.1
Contemporaneamente, Markowitz et al. mostraram resultados similares, demonstrando uma resposta imunológica satisfatória, com um ganho de CD4 de 74 e 83 células por mm3 após 4 e 12 semanas, respectivamente. Na semana 12, a queda da carga viral foi de 1,1 log.2
Referências Bibliográficas
1. Danner S, Carr A, Leonard J, Leahman L, Gudiol F Gonzalez J. A short term Study of the Safety, Pharmacokinetics and Efficacy of Ritonavir, an Inhibitor of HIV-1 Protease. N Engl J Med 1995;333:1528-33.
2. Markowitz M, Saag M, Powderly W, Hurley A, Hsu A, Valdes J. A Preliminary Study of Ritonavir, an Inhibitor of HIV-1 Protease, to Treat HIV Infection. N Engl J Med 1995;333:1534-9.
Solução Oral
A atividade de Ritonavir como monoterapia ou em combinação com inibidores da transcriptase reversa foi avaliada em 1.446 pacientes em dois estudos duplo-cegos randomizados.
Pacientes Avançados com Terapia Antirretroviral Prévia
O Estudo 247, randomizado, duplo-cego (com abertura de seguimento) foi conduzido em pacientes HIV infectados em terapia antirretroviral nos últimos 09 meses que apresentavam contagem celular média basal de CD4 menor ou igual a 100 células/mm3. Em cada paciente em regime de terapia antirretroviral (com até dois agentes antirretrovirais aprovados) foi administrado Ritonavir 600 mg duas vezes ao dia ou placebo.
O estudo ampliou-se com 1.090 pacientes, com contagem celular média basal de CD4 de 32 células/mm3. Após a demonstração dos benefícios clínicos da terapia com Ritonavir, todos os pacientes foram elegíveis a optar por Ritonavir durante o período de acompanhamento. A duração média da terapia duplocega com Ritonavir e placebo foi de 06 meses. A duração média do acompanhamento até a fase final do estudo aberto foi de 13,5 meses para pacientes randomizados com Ritonavir e 14 meses para os pacientes randomizados com placebo.
A incidência acumulada de progressão da doença ou morte durante a fase duplo-cega do Estudo 247 foi de 26% para os pacientes inicialmente randomizados com Ritonavir contra 42% dos pacientes inicialmente randomizados com o placebo. Essa diferença de porcentagem foi estatisticamente relevante. (veja Figura 01).
Figura 01: Tempo de Progressão da Doença ou Morte durante a fase duplo-cega do Estudo 247
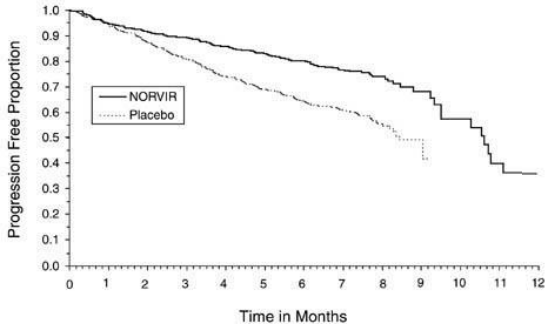
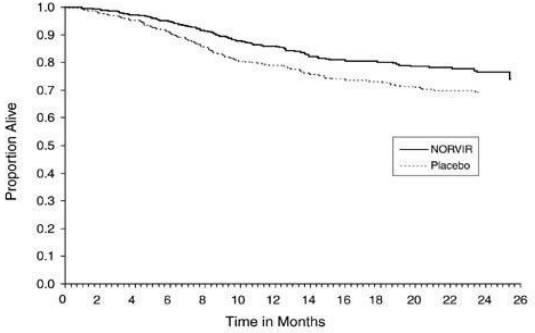
O acumulado de mortalidade no final da fase aberta de seguimento do Estudo 247 resultou em 18% para os pacientes inicialmente randomizados com Ritonavir e 26% para os pacientes inicialmente randomizados com placebo. Essa diferença na porcentagem foi estatisticamente relevante (veja Figura 02). Como a análise no final da fase aberta de seguimento incluiu pacientes que inicialmente estavam no grupo placebo e passaram a utilizar Ritonavir, os benefícios na sobrevida com Ritonavir não foram estimados com precisão.
Figura 02: Sobrevida dos pacientes no regime randomizado do Estudo 247
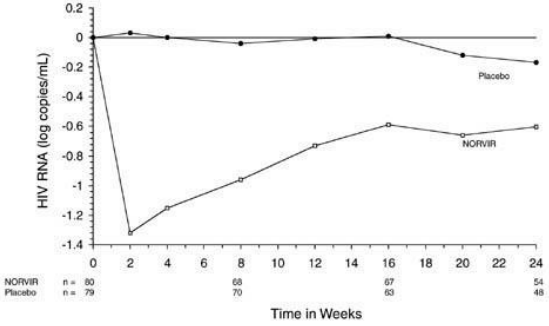
As Figuras 3 e 4 resumem as diferenças de contagem celular média basal de CD4 e de níveis de HIV RNA plasmático (cópias/mL) respectivamente, durante as primeiras 24 semanas da fase duplo-cega do Estudo 247.
Figura 03: Diferenças na contagem celular média basal de CD4 (células/mm3) durante a fase duplo-cega do Estudo 247
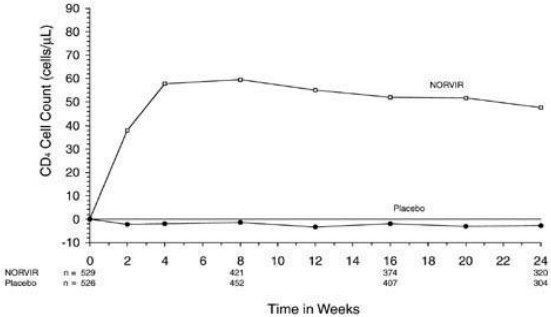
Figura 04: Diferenças no nível médio de HIV RNA plasmático (log cópias/mL) durante a fase duplo-cega do Estudo 247
Pacientes sem Terapia com antirretrovirais prévia
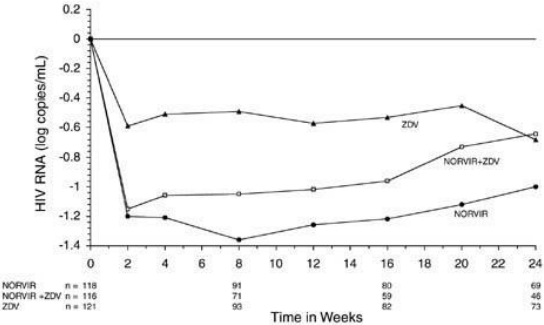
No Estudo 245, 356 pacientes HIV infectados sem terapia antirretroviral prévia - naive (média basal de CD4 = 364 células/mm3) foram randomizados para receber Ritonavir 600 mg duas vezes ao dia, zidovudina 200 mg três vezes ao dia ou uma combinação dessas drogas. A Figura 05 e 06 resumem a contagem celular média basal de CD4 e os níveis plasmáticos de HIV RNA (cópias/mL), respectivamente durante as primeiras 24 semanas da fase duplo-cega do Estudo 245.
Figura 05: Diferenças na contagem celular média basal de CD4 (células/mm3) durante o Estudo 245
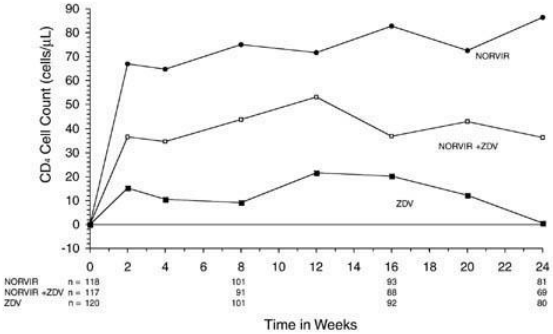
Figura 06: Diferenças nos níveis plasmáticos basais de HIV RNA durante o Estudo 245
Referências Bibliográficas
Caso haja interesse em conhecer as referências bibliográficas e/ou estudos clínicos disponíveis para este medicamento, por favor, entre em contato com a Central de Relacionamento AbbVie Line através do telefone 0800 022 2843.
Características Farmacológicas
O Ritonavir é um inibidor da protease do HIV, apresentando atividade contra o Vírus da Imunodeficiência Humana (HIV).
Farmacologia clínica
Mecanismo de ação
Ritonavir é um inibidor peptidomimético oral ativo das aspartil-proteases do HIV-1 e HIV-2. A inibição da protease do HIV torna a enzima incapaz de processar o precursor da poliproteína gag-pol, fazendo com que as partículas virais produzidas se tornem imaturas e, portanto, incapazes de iniciar um novo ciclo de infecção. O Ritonavir tem afinidade seletiva pela protease do HIV e pouca atividade inibitória diante da aspartil-protease humana. Pode ser utilizado também em conjunto com outros antirretrovirais da mesma classe com a finalidade de reduzir sua metabolização, diminuindo a dose necessária a cada tomada ou aumentando o intervalo entre as tomadas.
Atividade antiviral in vitro
Dados in vitro indicam que Ritonavir é ativo contra todas as cepas de HIV testadas em uma variedade de linhagens celulares humanas primárias e transformadas. A concentração da droga que inibe 50% e 90% da replicação viral in vitro é aproximadamente 0,02 mcM e 0,11 mcM, respectivamente. Potências similares foram observadas com cepas de HIV sensíveis e resistentes ao AZT. Estudos que avaliaram a citotoxicidade direta de Ritonavir em diversas linhagens celulares não mostraram toxicidade direta em concentrações de até 25 mcM, com um índice terapêutico resultante in vitro de pelo menos 1000.
Resistência
Isolados de HIV-1 resistentes ao Ritonavir foram selecionados in vitro. Os isolados resistentes demonstraram reduzir a susceptibilidade de Ritonavir e a análise genotípica desses isolados mostrou que a resistência foi primariamente atribuída a substituições específicas de aminoácidos na protease do HIV-1 nas posições 84 (IIe por Val), 82 (Val por Phe), 71 (Ala por Val) e 46 (Met por IIe). Alterações fenotípicas e genotípicas nos isolados de HIV de pacientes selecionados tratados com Ritonavir foram monitoradas em ensaios clínicos de Fase I/II. A análise genotípica e fenotípica em série indicou que a sensibilidade ao Ritonavir caiu de forma ordenada e gradual. As mutações iniciais ocorreram nas posições 82(Val por Ala/Phe), 54 (IIe por Val), 71 (Ala por Val/Thr) e 36 (IIe por Leu), seguidas pelas combinações de mutações em cinco posições adicionais específicas de aminoácidos. Cepas virais isoladas in vivo sem alteração na posição 82 não têm sensibilidade diminuída ao Ritonavir. A mutação na posição 82 parece ser necessária, mas não suficiente para conferir resistência fenotípica. A resistência fenotípica foi definida como uma diminuição ≥ 5 vezes na sensibilidade viral in vitro em relação ao basal. A relevância clínica das alterações genotípicas e fenotípicas associadas ao Ritonavir ainda não foi estabelecida.
Resistência cruzada com outros antirretrovirais
O potencial para resistência cruzada ao HIV entre inibidores de protease não foi completamente explorado. Portanto, não se conhece o efeito que Ritonavir terá na atividade de outros inibidores da protease administrados subsequentemente. Isolados de HIV obtidos em série de seis pacientes tratados com Ritonavir mostraram sensibilidade reduzida in vitro a este medicamento, mas não demonstraram redução correspondente da sensibilidade in vitro ao saquinavir quando comparada aos isolados basais. Entretanto, isolados de dois desses pacientes mostraram uma redução (8 vezes) na sensibilidade in vitro ao indinavir. Isolados de cinco pacientes também foram testados quanto à resistência cruzada ao amprenavir e nelfinavir; isolados de dois pacientes apresentaram diminuição da sensibilidade ao nelfinavir (12–14 vezes) e nenhuma ao amprenavir. A resistência cruzada entre Ritonavir e inibidores da transcriptase reversa é improvável, já que os alvos enzimáticos envolvidos são diferentes. Um isolado de HIV resistente á zidovudina (ZDV) testado in vitro manteve total sensibilidade ao Ritonavir.
Farmacocinética
Em um estudo de farmacocinética de dose única em indivíduos infectados pelo HIV do sexo masculino, em jejum, altos níveis do fármaco foram encontrados e mantidos por várias horas após administração oral de 100 mg, 200 mg, 400 mg, 600 mg, 800 mg ou 1000 mg de Ritonavir. A área sob a curva (AUC) da concentração versus tempo variou de 3,92 a 123 mcg.h/mL, respectivamente e a Cmáx variou de 0,416 a 12,7 mcg/mL. A farmacocinética do Ritonavir foi dose-dependente e aumentos maiores do que o proporcional na AUC e Cmáx com aumento de dose foram relatados.
O Tmáx permaneceu constante por aproximadamente 3 horas com o aumento de dose. A depuração renal foi, em média, inferior a 0,1 L/h e relativamente constante na faixa de dosagem. Como não há formulação parenteral de Ritonavir, a biodisponibilidade absoluta não foi determinada.
Após uma dose única de 600 mg sob condições de plenitude gástrica, as formulações em cápsula gelatinosa mole de 100 mg (n=57) e em solução oral (n=18) produziram AUCs de 121,7 + 53,8 mcg.h/mL e 129,0 + 39,3 mcg.h/mL (média + desvio padrão), respectivamente. Em comparação à ingestão em jejum, a extensão de absorção da cápsula gelatinosa mole foi 12% maior quando administrada com uma refeição. Quando a formulação líquida foi dada sob condições de jejum, as concentrações de pico de Ritonavir aumentaram 28% em relação às condições de plenitude.
A farmacocinética do Ritonavir durante regimes de múltiplas doses foram estudadas em voluntários adultos HIV-positivos em condições de plenitude gástrica. Sob condições de múltiplas doses, o acúmulo de Ritonavir é ligeiramente menor do que o previsto a partir da dose única devido a um aumento relacionado ao tempo e à dose na depuração aparente (Cl/F). Foi observado que concentrações mínimas de Ritonavir diminuem com o tempo, possivelmente devido à indução enzimática, atingindo níveis estáveis após 2 semanas. No estado de equilíbrio, com uma dose de 600 mg duas vezes ao dia, foram observados valores de Cmáx e Cmín de 11,2 e 3,7 mcg/mL, respectivamente. O t½ de Ritonavir foi de aproximadamente 3 a 5 horas. A depuração aparente no estado de equilíbrio em pacientes tratados com 600 mg duas vezes ao dia foi em média 8,8 ± 3,2 L/h.
Não foram observadas diferenças clinicamente significantes na AUC ou Cmáx entre homens e mulheres. Os parâmetros farmacocinéticos do Ritonavir não foram significativamente associados à perda de peso ou massa corpórea magra.
O volume aparente de distribuição (VB/F) do Ritonavir é de aproximadamente 0,41 ± 0,25 L/kg após uma dose única de 600 mg. A ligação do Ritonavir a proteínas no plasma humano foi de aproximadamente 98 a 99%. O Ritonavir se liga à alfa-1 glicoproteína ácida (AAG) e à albumina sérica humana (HSA) com afinidades comparáveis. A ligação a proteínas plasmáticas total é constante na faixa de concentração de 1 a 100 mcg/mL.
Estudos de distribuição tecidual em ratos com Ritonavir marcado com 14C demonstraram que o fígado, as suprarrenais, o pâncreas, os rins e a tireoide tinham as maiores concentrações de Ritonavir. Uma relação tecido-plasma de aproximadamente 1, medido nos gânglios linfáticos de ratos, sugere que o Ritonavir é distribuído no tecido linfático. O Ritonavir penetra de maneira mínima no cérebro.
Observou-se que o Ritonavir é extensamente metabolizado pelo sistema do citocromo hepático P450, principalmente pela isoenzima CYP3A e em menor extensão pela CYP2D6. Estudos em animais, assim como experimentos in vitro com microssomos hepáticos humanos, indicam que o Ritonavir sofre principalmente metabolismo oxidativo. Cinco metabólitos de Ritonavir foram identificados no homem. O metabólito da oxidação isopropiltiazólico (M-2) é o principal metabólito e tem atividade antiviral similar à da substância precursora. Entretanto, a AUC do metabólito M-2 foi aproximadamente 3% da AUC da substância precursora.
Estudos em humanos com Ritonavir marcado radioativamente demonstraram que sua eliminação se faz principalmente pela via hepatobiliar; aproximadamente 86% da substância marcada foi recuperada nas fezes. Nestes estudos, a via renal de eliminação não foi observada como via de eliminação importante do Ritonavir.
Efeitos no Eletrocardiograma
O intervalo QTcF foi avaliado em um estudo controlado cruzado, randomizado, placebo e ativo (moxifloxacina 400 mg/uma vez ao dia), com 45 adultos sadios, com 10 medidas durante 12 horas no Dia 3. A média de diferença máxima (limite de confiança superior a 95%) no QTcF do placebo foi de 5,5 (7,6) mseg para Ritonavir 400 mg duas vezes ao dia. A exposição ao Ritonavir no Dia 3 foi aproximadamente 1,5 vezes maior que a observada com a dose de 600 mg duas vezes ao dia no estado de equilíbrio. Nenhum voluntário teve um aumento na QTcF > 60 mseg da baseline ou um intervalo QTcF que excedesse o limite clinicamente relevante de 500 mseg.
Um discreto prolongamento no intervalo PR também foi verificado em voluntários recebendo Ritonavir durante o mesmo estudo no Dia 3. O intervalo PR máximo foi de 252 mseg e não houve bloqueio cardíaco de segundo ou terceiro graus.
Populações Especiais
Insuficiência renal
Atualmente não há dados específicos sobre esta população de pacientes. Entretanto, devido à alta ligação protéica de Ritonavir é pouco provável que o Ritonavir seja significativamente removido por hemodiálise ou diálise peritoneal.
Insuficiência hepática
Em seis indivíduos adultos, com doença hepática leve, infectados pelo HIV, recebendo 400 mg de Ritonavir duas vezes ao dia, a exposição ao Ritonavir foi semelhante em comparação com os indivíduos controles recebendo 500 mg de Ritonavir duas vezes ao dia. Os resultados indicam que não é necessário ajuste de doses em pacientes com doença hepática leve. Dados adequados de farmacocinética não estão disponíveis para pacientes com doença hepática moderada. A ligação protéica do Ritonavir não foi afetada de modo estatisticamente significativo pela doença hepática leve e moderada.
Pacientes pediátricos
Farmacocinética no estado de equilíbrio foi avaliada em 37 pacientes com idades entre 2 e 14 anos infectados por HIV, que recebiam doses variando entre 250 mg/m2 de superfície corporal e 400 mg/m2 de superfície corporal duas vezes ao dia, no Grupo de Estudo de Pacientes Pediátricos com AIDS do estudo 310 (PACTG), e em 41 pacientes com idades entre 1 mês e 2 anos, infectados por HIV, em doses de 350 e 450 mg/m2 duas vezes ao dia, no estudo 345 em PACTG.
A depuração de Ritonavir oral no estado de equilíbrio foi aproximadamente 1,5 a 1,7 vezes mais rápida em pacientes pediátricos do que em adultos. As concentrações de Ritonavir em pacientes pediátricos, maiores de 2 anos, obtidas após 350 a 400 mg/m2 duas vezes ao dia, foram comparáveis àquelas obtidas em adultos recebendo 600 mg (aproximadamente 330 mg/m2) duas vezes ao dia. As seguintes observações foram feitas a respeito das concentrações do Ritonavir após a administração de 350 ou 450 mg/m2, duas vezes ao dia, em crianças menores de 2 anos de idade. Exposições mais elevadas do Ritonavir não foram evidentes com 450 mg/m2 duas vezes ao dia comparado a 350 mg/m2 duas vezes ao dia. As concentrações mais baixas de Ritonavir eram um pouco menores do que aquelas obtidas nos adultos que receberam 600 mg duas vezes ao dia.
A área sob a curva de concentração plasmática de Ritonavir/tempo e menores concentrações obtidas após a administração de 350 ou 450 mg/m2, duas vezes ao dia, em crianças menores de 2 anos, foram aproximadamente 16% e 60% mais baixas, respectivamente, do que aquelas obtidas em adultos que receberam 600 mg duas vezes ao dia.
Dados de segurança pré-clínica
Toxicidade Aguda, Subaguda e Crônica
O Ritonavir apresenta baixos índices de toxicidade aguda quando administrado oralmente. A DL50 foi determinada como sendo maior do que 2500 mg/kg, tanto em ratos quanto em camundongos. Os sinais de toxicidade com altas doses nas duas espécies incluíram diminuição da atividade, ataxia, dispneia e tremores. Em geral, os sinais de toxicidade foram aparentes de 1 a 3 dias após a administração. Nenhuma mudança morfológica bruta foi vista em necropsia de ratos depois de um período de 2 semanas de observação.
Os estudos de toxicidade com doses repetidas nos animais identificaram como principais órgãos-alvo: o fígado, a glândula tireoide, retina e o rim. Alterações hepáticas envolveram elementos hepatocelular, biliar e fagocíticos e foram acompanhados de aumentos nas enzimas hepáticas. Hipertrofia no epitélio pigmentado da retina (RPE) e degeneração da retina foram observados nos roedores nos estudos conduzidos com Ritonavir, mas não foram observados em cachorros. Evidências ultraestruturais sugeriram que essas alterações na retina em roedores podem ser secundárias a fosfolipidose. Entretanto, três experiências clínicas de fase II não revelaram evidências claras de alterações na retina por indução do fármaco em humanos. Alterações relacionadas à glândula tireoide incluíram hipertrofia das células foliculares, diminuição dos níveis séricos de tiroxina (T4) e/ou aumento dos níveis séricos de TSH. Todas as alterações tireoidianas foram reversíveis após a descontinuação do fármaco. Investigações clínicas em humanos não revelaram alterações clínicas significantes nos testes das funções da tireoide. Alterações renais, incluindo degeneração tubular, inflamação crônica e proteinúria, foram observadas em ratos e foram atribuídas a doenças espécieespecíficas espontâneas. Além disso, não foram observadas alterações renais clinicamente significantes nas experiências clínicas.
Exclusivo Comprimido
A alimentação reduz ligeiramente a biodisponibilidade de Ritonavir comprimidos. A administração de uma dose única de 100 mg de Ritonavir comprimidos com uma alimentação moderada em gordura (857 Kcal, 31% calorias em gordura) ou uma alimentação rica em gordura (907 Kcal, 52% calorias em gordura) foi associado com uma diminuição de 20-23% na AUC e na Cmáx de Ritonavir.
O Ritonavir é um pó branco ou com leve cor marrom, de sabor metálico amargo. É facilmente solúvel em metanol e etanol, solúvel em isopropanol e praticamente insolúvel em água.
O Ritonavir é chamado quimicamente de éster 5-tiazolilmetílico, [5S- (5R*,8R*,10R*,11R*)] do ácido 10-hidroxi-2-metil-5-(1-metiletil)-1-[2-(1- metiletil)-4-tiazolil]-3,6-dioxo-8,11-bis(fenilmetil)-2,4,7,12-tetraazatridecan-13- óico. Sua fórmula molecular é C37H48N6O5S2 e seu peso molecular é 720,95.
Exclusivo Solução Oral
Carcinogênese e mutagênese
O Ritonavir não foi mutagênico ou clastogênico nos estudos in vitro e in vivo que incluíram ensaios de mutação reversa Ames usando S. typhimurium e E. coli, ensaios com linfoma de camundongos, teste de micronúcleo em camundongos e ensaio de aberração cromossômica em linfócitos humanos. Além disso, estudos de carcinogenicidade em ratos e camundongos indicaram que o Ritonavir não tem ação carcinogênica direta nas dosagens testadas. Uma incidência maior de adenoma hepatocelular foi encontrada em camundongos machos recebendo alta dose de 200 mg/kg/dia. Tais respostas tumorais em fígado de camundongos associadas à compostos não-genotóxicos são consideradas de pequena relevância para a resposta do fígado humano.