 Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)
Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)Qual a ação da substância do Otezla?
Características Farmacológicas
Propriedades farmacodinâmicas
Grupo farmacoterapêutico: imunossupressores, imunossupressores seletivos.
Código ATC: L04AA32.
Mecanismo de ação
O Apremilast, uma pequena molécula oral inibidora da fosfodiesterase 4 (PDE4), atua intracelularmente de modo a modular uma rede de mediadores pro-inflamatórios e anti-inflamatórios. A PDE4 é uma PDE específica da adenosina monofosfato cíclica (AMPc) e é a PDE dominante nas células inflamatórias. A inibição da PDE4 aumenta os níveis intracelulares de AMPc que, por sua vez, hiporregulam a resposta inflamatória ao modularem a expressão do TNF-α, IL-23, IL-17 e de outras citocinas inflamatórias. A AMPc modula também os níveis de citocinas anti-inflamatórias tais como a IL-10. Este mediadores pró e anti-inflamatórios têm sido implicados na artrite psoriática e na psoríase.
Efeitos farmacodinâmicos
Nos estudos clínicos em doentes com artrite psoriática, o Apremilast modulou significativamente, mas não inibiu na sua totalidade, os níveis de proteínas plasmáticas de IL-1α, IL-6, IL-8, MCP-1, MIP-1β, MMP-3 e TNF-α. Após 40 semanas de tratamento com Apremilast, verificou-se uma diminuição dos níveis de proteínas plasmáticas de IL-17 e IL-23, e um aumento de IL-10. Nos ensaios clínicos em doentes com psoríase, o Apremilast diminuiu a espessura epidérmica da lesão cutânea, a infiltração de células inflamatórias e a expressão de genes pró-inflamatórios, inclusive os responsáveis pela sintetase do óxido nítrico induzível (iNOS), IL-12/IL-23p40, IL-17A, IL-22 e IL-8.
O Apremilast administrado em doses até 50 mg BID não prolongou o intervalo QT em indivíduos saudáveis.
Experiência com ensaios clínicos
Artrite psoriática
A segurança e eficácia do Apremilast foram avaliadas em 3 estudos multicêntricos, aleatorizados, em dupla ocultação, controlados por placebo (estudos PALACE 1, PALACE 2 e PALACE 3), com desenho semelhante, em doentes adultos com PsA ativa (≥ 3 articulações edemaciadas e ≥ 3 articulações dolorosas) apesar de tratamento anterior com DMARDs de pequenas moléculas ou biológicos. Um total de 1493 doentes foi aleatorizado e tratado com placebo, Apremilast 20 mg ou Apremilast 30 mg administrado três vezes por dia, por via oral.
Os doentes nestes estudos tinham diagnóstico de PsA há pelo menos 6 meses. O PALACE 3 exigia ainda uma lesão psoriática cutânea qualificável (com pelo menos 2 cm de diâmetro). O Apremilast foi utilizado em monoterapia (34,8%) ou em associação com doses estáveis de DMARDs de pequenas moléculas (65,2%).
Os doentes receberam Apremilast em associação com um ou mais dos seguintes:
Metotrexato (MTX, ≤ 25 mg/semana; 54,5%), sulfasalazina (SSZ, ≤ 2 g/dia; 9,0%), e leflunomida (LEF, ≤ 20 mg/dia; 7,4%). Não foi permitido tratamento concomitante com DMARDs biológicos, incluindo bloqueadores do TNF.
Foram recrutados para os 3 estudos doentes com cada subtipo de PsA, incluindo poliartrite simétrica (62,0%), oligoartrite assimétrica (26,9%), artrite articular interfalângica distal (DIP) (6,2%), artrite mutilante (2,7%) e espondilite predominante (2,1%). Foram recrutados doentes com entesopatia pré-existente (63%) ou com dactilite pré-existente (42%). Um total de 76,4% de doentes tinha sido tratado previamente apenas com DMARDs de pequenas moléculas e 22,4% dos doentes tinham sido tratados previamente com DMARDs biológicos, o que inclui 7,8% com um insucesso terapêutico anterior com um DMARD biológico. A duração mediana da PsA foi de 5 anos.
Com base no desenho do estudo, os doentes cujo número de articulações dolorosas e edemaciadas não tivessem melhorado pelo menos 20% eram considerados não respondedores à semana 16. Os doentes a fazerem placebo que fossem considerados não respondedores eram novamente aleatorizados numa razão de 1:1, em ocultação, de modo a receberem Apremilast 20 mg duas vezes por dia ou 30 mg duas vezes por dia. À semana 24, todos os restantes doentes tratados com placebo passaram para o Apremilast 20 mg ou 30 mg BID.
O parâmetro de avaliação primário foi a percentagem de doentes que atingiram resposta 20 do American College of Rheumatology (ACR) à semana 16.
O tratamento com Apremilast resultou numa melhoria significativa dos sinais e sintomas da PsA, conforme avaliado pelos critérios da resposta ACR 20 comparativamente ao placebo à semana 16. A proporção de doentes com ACR 20/50/70 (as respostas nos estudos PALACE 1, PALACE 2 e PALACE 3 assim como os dados agrupados dos estudos PALACE 1, PALACE 2 e PALACE 3) para o Apremilast 30 mg duas vezes por dia à semana 16, encontram-se na Tabela 3. As respostas ACR 20/50/70 mantiveram-se na semana 24.
Entre os doentes inicialmente aleatorizados para o tratamento com Apremilast 30 mg duas vezes ao dia, as taxas de resposta ACR 20/50/70 mantiveram-se até à semana 52 nos estudos agrupados PALACE 1, PALACE 2 e PALACE 3.
Tabela 3: Proporção de doentes com respostas ACR nos estudos PALACE 1, PALACE 2 e PALACE 3 e nos estudos agrupados à Semana 16
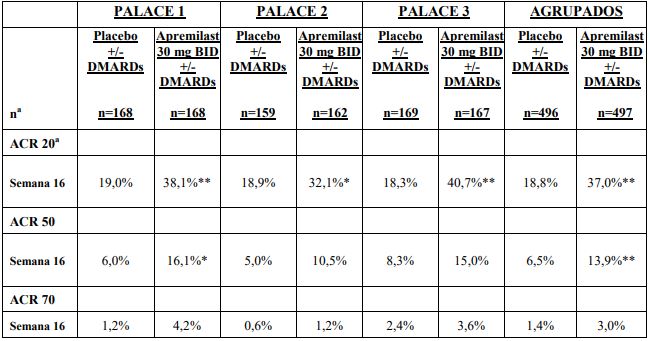
* p ≤ 0,01 para o Apremilast vs. placebo.
** p ≤ 0,001 para o Apremilast vs. placebo.
a n Corresponde ao número de doentes aleatorizados e tratados.
Figura 1: Proporção de respondedores ACR 20/50/70 até à semana 52 na análise agrupada dos estudos PALACE 1, PALACE 2 e PALACE 3 (NRI*)
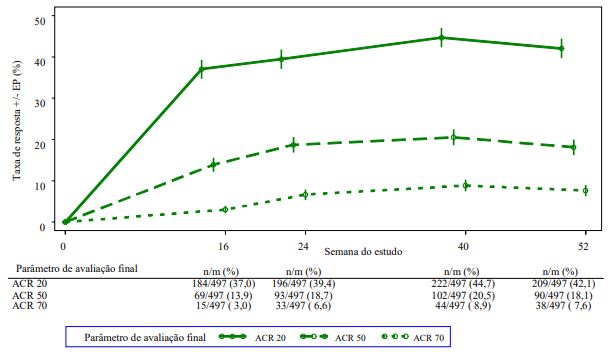
*NRI: Imputação a não respondedores. Os indivíduos que descontinuaram precocemente antes do ponto temporal e os indivíduos que não tiveram dados suficientes para uma determinação definitiva do estado de resposta no ponto temporal são contabilizados como não respondedores.
Entre os 497 doentes inicialmente aleatorizados para o Apremilast 30 mg duas vezes por dia, 375 (75%) doentes ainda estavam a fazer este tratamento na semana 52. Nestes doentes, as respostas ACR 20/50/70 à semana 52 foram 57%, 25% e 11%, respetivamente.
As respostas observadas no grupo tratado com Apremilast foram semelhantes nos doentes medicados e não medicados com DMARDs concomitantemente, incluindo MTX.
Os doentes previamente tratados com DMARDs ou com biológicos que receberam Apremilast atingiram uma maior resposta ACR 20 à semana 16 do que os doentes que estavam a receber placebo.
Observaram-se respostas ACR semelhantes em doentes com diferentes subtipos de PsA, incluindo DIP. O número de doentes com subtipos de artrite mutilante e de espondilite predominante foi demasiado pequeno para permitir uma avaliação significativa.
No PALACE 1, PALACE 2 e PALACE 3 as melhorias da proteína C reativa (CRP) na Escala de Atividade da Doença (DAS) 28 e na proporção de doentes que atingiu critérios de resposta modificados para a PsA (PsARC) foram maiores no grupo do Apremilast em comparação com o placebo à semana 16 (valor de p nominal p < 0,0004; valor de p < 0,0017, respetivamente). Estas melhorias mantiveram-se à semana 24. Entre os doentes que permaneceram no tratamento com Apremilast para o qual tinham sido aleatorizados no início do estudo, a pontuação DAS28(CRP) e a resposta PsARC mantiveram-se até à semana 52.
Às semanas 16 e 24 observaram-se melhorias nos parâmetros da atividade periférica característica da artrite psoriática (por ex. número de articulações edemaciadas, número de articulações dolorosas dactilites e entesites) e nas manifestações cutâneas da psoríase nos doentes tratados com Apremilast.
Entre os doentes que permaneceram no tratamento com Apremilast para o qual tinham sido aleatorizados no início do estudo, estas melhorias mantiveram-se até à semana 52.
Função física e qualidade de vida relacionada com a saúde
Os doentes tratados com Apremilast demonstraram uma melhoria estatisticamente significativa da função física, conforme avaliação com o índice de incapacidade do questionário de avaliação da saúde (HAQ-DI) das alterações desde o início do estudo, comparativamente ao placebo na semana 16 no PALACE 1, PALACE 2 e PALACE 3 e nos estudos agrupados (Tabela 4). A melhoria nas pontuações do HAQ-DI manteve-se à semana 24.
Entre os doentes que foram inicialmente aleatorizados para tratamento com Apremilast 30 mg duas vezes por dia, a alteração na pontuação do HAQ-DI entre o início do estudo e a semana 52 foi de -0,333 no grupo do Apremilast 30 mg duas vezes por dia, numa análise agrupada da fase sem ocultação dos estudos PALACE 1, PALACE 2 e PALACE 3.
Nos estudos PALACE 1, PALACE 2 e PALACE 3, foram demonstradas melhorias significativas da qualidade de vida relacionada com a saúde, avaliadas pelas alterações desde o início do estudo no domínio da função física (PF) da versão 2 do Questionário de Saúde - Versão Curta, (SF-36v2) e nas pontuações da Avaliação Funcional da Fadiga na Terapêutica da Doença Crónica (FACIT-fatigue) nos doentes tratados com Apremilast em comparação com o placebo à semana 16 e 24. Entre os doentes que permaneceram no tratamento com Apremilast, para o qual tinham sido inicialmente aleatorizados no início do estudo, as melhorias na função física e FACIT- fatigue mantiveram-se até à semana 52.
Psoríase
A segurança e eficácia do Apremilast foram avaliadas em dois estudos multicêntricos, aleatorizados, em dupla ocultação, e controlados por placebo (estudos ESTEEM 1 e ESTEEM 2) que recrutaram um total de 1257 doentes com psoríase em placas moderada a grave, com um envolvimento da área de superfície corporal (BSA) ≥ 10%, uma pontuação do Índice de Gravidade e Extensão da Psoríase (PASI) ≥ 12, Avaliação Global pelo Médico (sPGA) ≥ 3 (moderada ou grave) e que eram candidatos para fototerapia ou para terapêutica sistémica.
Estes estudos tiveram um desenho semelhante até à semana 32. Em ambos os estudos, os doentes foram aleatorizados numa razão de 2:1 para Apremilast 30 mg BID ou placebo durante 16 semanas (fase controlada por placebo) e entre as semanas 16-32 todos os doentes receberam Apremilast 30 mg BID (fase de manutenção). Durante a fase aleatorizada de descontinuação do tratamento (semanas 32- 52), os doentes originalmente aleatorizados para o Apremilast que atingiram uma redução de pelo menos 75% na sua pontuação PASI (PASI-75) (ESTEEM 1) ou uma redução de 50% na sua pontuação PASI (PASI-50) (ESTEEM 2) foram novamente aleatorizados na semana 32 para placebo ou Apremilast 30 mg BID. Os doentes que foram novamente aleatorizados para o placebo e que perderam a resposta PASI-75 (ESTEEM 1) ou que perderam 50% de melhoria PASI na semana 32, em comparação com o início do estudo (ESTEEM 2), foram novamente tratados com Apremilast 30 mg BID.
Os doentes que não atingiram a resposta PASI estabelecida até à semana 32, ou que foram inicialmente aleatorizados para placebo, permaneceram com Apremilast até à semana 52. A utilização de corticosteroides tópicos de baixa potência na face, axilas e virilhas, champô de alcatrão de carvão e/ou preparações com ácido salicílico para o couro cabeludo foi permitida durante os estudos. Adicionalmente, na semana 32, aos indivíduos que não atingiram uma resposta PASI-75 no ESTEEM 1 ou uma resposta PASI-50 no ESTEEM 2, foi permitido utilizar terapêuticas tópicas para a psoríase e/ou fototerapia além do tratamento com Apremilast 30 mg BID.
Em ambos os estudos, o parâmetro de avaliação primário foi a proporção de doentes que atingiu uma PASI-75 na semana 16. O principal parâmetro de avaliação secundário foi a proporção de doentes que atingiu uma pontuação sPGA de limpo (0) ou quase limpo (1) na semana 16.
A pontuação PASI média no início do estudo foi 19,07 (mediana de 16,80) e a proporção de doentes com uma pontuação sPGA de 3 (moderado) e 4 (grave) no início do estudo foi de 70,0% e 29,8%, respetivamente, com um envolvimento médio de 25,19% de BSA (mediana de 21,0%). Cerca de 30% de todos os doentes tinha recebido fototerapia prévia e 54% tinha recebido terapêutica sistémica convencional e/ou biológica prévia para o tratamento da psoríase (incluindo insucessos terapêuticos), tendo 37% recebido terapêutica sistémica convencional e 30% recebido terapêutica biológica prévias. Cerca de um terço dos doentes não recebeu fototerapia, terapêutica sistémica convencional ou terapêutica biológica prévias. Dezoito por cento dos doentes tinham antecedentes de artrite psoriática.
A proporção de doentes que atingiu respostas PASI 50,75 e 90 e uma pontuação sPGA de limpo (0) ou quase limpo (1) é apresentada na Tabela 4. O tratamento com Apremilast resultou numa melhoria significativa da psoríase em placas moderada a grave, conforme demonstrado pela proporção de doentes com resposta PASI-75 à semana 16, em comparação com o placebo. A melhoria clínica medida pelas respostas sPGA, PASI-50 e PASI-90 foi também demonstrada à semana 16. Adicionalmente, o Apremilast demonstrou benefício terapêutico em inúmeras manifestações da psoríase incluindo prurido, onicopatias, envolvimento do couro cabeludo e medidas ao nível da qualidade de vida.
Tabela 4: Resposta clínica à semana 16 nos estudos ESTEEM 1 e ESTEEM 2 (FAS a LOCFb )
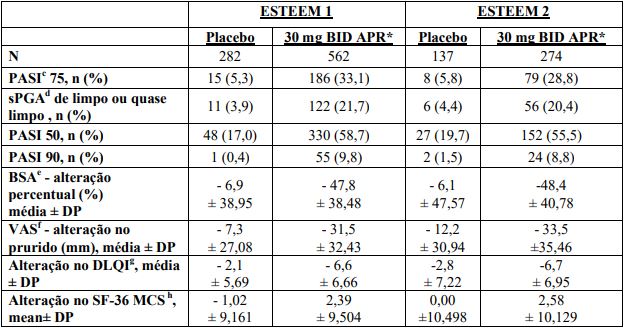
* p< 0,0001 para o Apremilast vs placebo, exceto para PASI 90 e alteração do SF-36 MCS no ESTEEM 2 nos quais p=0,0042 e p=0,0078, respetivamente.
a FAS = Conjunto de análise total.
b LOCF= Última observação efetuada.
c PASI = Índice de Gravidade.
e Extensão da Psoríase.
d sPGA = Avaliação Global pelo Médico.
e BSA = Área de Superfície Corporal
f VAS = Escala Visual Analógica; 0 = melhor, 100 = pior.
g DLQI = Índice Dermatológico de Qualidade de Vida; 0 = melhor, 30 = pior.
h SF-36 MCS = versão curta com 36 items do Questionário de Saúde - do Estudo dos Resultados Médicos, Resumo da Componente Mental.
O benefício clínico do Apremilast foi demonstrado em inúmeros subgrupos definidos pelos dados demográficos no início do estudo e pelas características clínicas da doença no início do estudo (incluindo a duração da psoríase enquanto doença e doentes com antecedentes de artrite psoriática). O benefício clínico do Apremilast foi também demonstrado independentemente da utilização prévia de medicação para a psoríase e da resposta a tratamentos prévios para a psoríase. As taxas de resposta foram semelhantes para todos os intervalos de peso.
A resposta ao Apremilast foi rápida, com melhorias significativamente superiores nos sinais e sintomas da psoríase, incluindo o PASI, desconforto/dor cutâneos e prurido, em comparação com o placebo na semana 2. Em geral, as respostas PASI foram atingidas à semana 16 e mantiveram-se até à semana 32.
Em ambos os estudos, a melhoria percentual média no PASI desde o início do estudo permaneceu estável durante a Fase Aleatorizada de Descontinuação do Tratamento para os doentes novamente aleatorizados para o Apremilast na semana 32.
Tabela 5: Persistência do efeito em indivíduos aleatorizados para APR 30 BID à semana 0 e novamente aleatorizados para APR 30 BID da semana 32 à semana 52
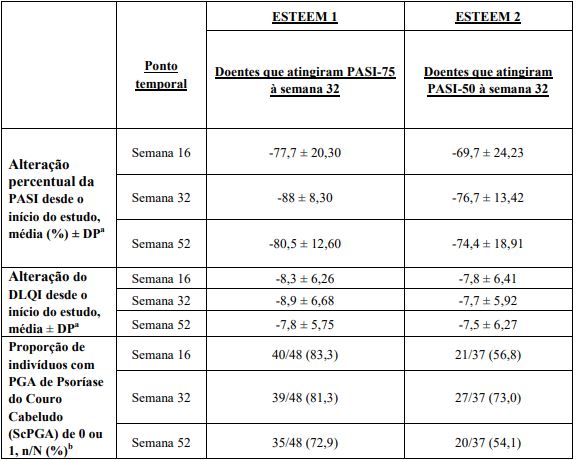
a Inclui indivíduos novamente aleatorizados para APR 30 BID à semana 32 com um valor basal e pós basal na semana de estudo avaliada.
b N baseia-se em indivíduos com psoríase do couro cabeludo moderada ou superior no início do estudo que foram novamente aleatorizados para APR 30 BID à semana 32. Os indivíduos com dados em falta foram considerados como não respondedores.
No estudo ESTEEM 1, cerca de 61% dos doentes que foram novamente aleatorizados para Apremilast na semana 32 tiveram uma resposta PASI-75 na semana 52. Dos doentes com pelo menos resposta PASI-75 que foram novamente aleatorizados para placebo à semana 32 durante uma Fase Aleatorizada de Descontinuação do Tratamento, 11,7% foram respondedores PASI-75 na semana 52. O tempo mediano até perda da resposta PASI-75 nos doentes que foram novamente aleatorizados para placebo foi de 5,1 semanas.
No estudo ESTEEM 2, cerca de 80,3% dos doentes novamente aleatorizados para Apremilast na semana 32 tiveram uma resposta PASI-50 à semana 52. Dos doentes com pelo menos resposta PASI50 novamente aleatorizados para placebo na semana 32, 24,2% foram respondedores PASI-50 na semana 52. O tempo mediano até perda de 50% da sua melhoria PASI na semana 32 foi de 12,4 semanas.
Após a descontinuação aleatorizada da terapêutica na semana 32, aproximadamente 70% dos doentes no estudo ESTEEM 1 e 65,6% dos doentes no estudo ESTEEM 2, voltaram a atingir respostas PASI75 (ESTEEM 1) ou PASI-50 (ESTEEM 2) após o reinício do tratamento com Apremilast. Devido ao desenho do estudo, a duração do segundo tratamento foi variável, oscilando entre 2,6 a 22,1 semanas.
No estudo ESTEEM 1, aos doentes aleatorizados para Apremilast no início do estudo que não atingiram uma resposta PASI-75 ao fim da semana 32, foi permitida a utilização concomitante de terapêuticas tópicas e/ou fototerapia UVB entre as semanas 32 a 52. Destes doentes, 12% atingiram resposta PASI-75 na semana 52 com Apremilast mais tratamento tópico e/ou fototerapia.
Nos estudos ESTEEM 1 e ESTEEM 2, na semana 16, foram observadas melhorias significativas (diminuições) da psoríase ungueal, avaliadas pela alteração percentual média do Índice de Severidade da Psoríase Ungueal (NAPSI) desde o início do estudo, nos doentes que estavam a receber Apremilast em comparação com os doentes tratados com placebo (p< 0,0001 e p=0,0052, respetivamente). Foram observadas outras melhorias na psoríase ungueal na semana 32, nos doentes em tratamento continuado com Apremilast.
Nos estudos ESTEEM 1 e ESTEEM 2, foram observadas melhorias significativas na psoríase do couro cabeludo de intensidade pelo menos moderada (≥3), medida pela proporção de doentes que atingiram uma Avaliação Global da Psoríase do Couro Cabeludo pelo Médico (ScPGA) de limpo (0) ou mínimo (1) à semana 16, nos doentes a receberem Apremilast em comparação com os doentes tratados com placebo (p< 0,0001 para ambos os estudos). Na generalidade, as melhorias mantiveram-se nos indivíduos que foram novamente aleatorizados para Apremilast desde a semana 32 até à semana 52.
Nos estudos ESTEEM 1 e ESTEEM 2, foram demonstradas melhorias significativas na qualidade de vida avaliada pelo Índice Dermatológico de Qualidade de Vida (DLQI) e pelo SF-36v2MCS nos doentes a receberem Apremilast em comparação com os doentes tratados com placebo (Tabela 4). As melhorias no DLQI mantiveram-se até à semana 52 nos indivíduos que foram novamente aleatorizados para Apremilast na semana 32 (Tabela 5). Adicionalmente, no estudo ESTEEM 1, atingiu-se uma melhoria significativa no Índice do Questionário sobre Limitações no Trabalho (WLQ-25) nos doentes a receberem Apremilast em comparação com o placebo.
Propriedades farmacocinéticas
Absorção
O Apremilast é bem absorvido com uma biodisponibilidade oral absoluta de aproximadamente 73%, com concentrações plasmáticas máximas (Cmax) a ocorrerem num tempo mediano (tmax) de aproximadamente 2,5 horas. A farmacocinética do Apremilast é linear, com um aumento proporcional à dose da exposição sistémica no intervalo de dose dos 10 aos 100 mg por dia. A acumulação é mínima quando o Apremilast é administrado uma vez por dia e de cerca de 53% em indivíduos saudáveis e 68% em doentes com psoríase quando administrado duas vezes pordia. A coadministração com alimentos não altera a biodisponibilidade, portanto, o Apremilast pode ser administrado com ou sem alimentos.
Distribuição
A ligação do Apremilast às proteínas plasmáticas humanas é de aproximadamente 68%. O volume de distribuição (Vd) aparente médio é de 87 l, indicativo de distribuição extravascular.
Biotransformação
O Apremilast é extensivamente metabolizado tanto por vias mediadas ou não mediadas pela CYP, incluindo oxidação, hidrólise e conjugação, o que sugere que é pouco provável que a inibição de uma única via de depuração possa causar uma interação medicamentosa acentuada. O metabolismo oxidativo do Apremilast é maioritariamente mediado pela CYP3A4, com pequenas contribuições da CYP1A2 e da CYP2A6. O Apremilast é o principal componente em circulação após a administração oral. O Apremilast é sujeito a um metabolismo extenso com apenas 3% e 7% do composto original administrado recuperado na urina e nas fezes, respetivamente. O principal metabolito inativo circulante é o conjugado glucoronídeo de O-demetil Apremilast (M12). A exposição ao Apremilast diminui quando administrado concomitantemente com rifampicina, um potente indutor da CYP3A4, o que é consistente com o facto do Apremilast ser um substrato da CYP3A4.
In vitro, o Apremilast não é um inibidor ou indutor das enzimas do citocromo P450. Por conseguinte, é improvável que a coadministração de Apremilast com substratos das enzimas CYP afete a depuração e exposição de substâncias ativas metabolizadas pelas enzimas CYP.
In vitro, o Apremilast é um substrato e um inibidor fraco da glicoproteína P (CI50>50 µM), contudo, não é de prever que ocorram interações medicamentosas clinicamente relevantes.
In vitro, o Apremilast tem pouco ou nenhum efeito inibitório (CI50>10 µM) no transportador de aniões orgânicos (OAT)1 e no OAT3, no transportador de catiões orgânicos (OCT)2, no polipéptido transportador de aniões orgânicos (OATP)1B1 e no OATP1B3 ou na proteína de resistência do cancro da mama (BCRP) e não é um substrato para estes transportadores. Como tal, é improvável que ocorram interações medicamentosas clinicamente relevantes quando o Apremilast é coadministrado com fármacos que são substratos ou inibidores destes transportadores.
Eliminação
A depuração plasmática do Apremilast é, em média, de cerca de 10 l/h em indivíduos saudáveis, com uma semivida de eliminação terminal de aproximadamente 9 horas. Após a administração oral de Apremilast radiomarcado, cerca de 58% e 39% da radioatividade é recuperada na urina e nas fezes, respetivamente, com cerca de 3% e 7% da dose radioativa recuperada sob a forma de Apremilast na urina e nas fezes, respetivamente.
Doentes idosos
O Apremilast foi estudado em indivíduos jovens e idosos saudáveis. A exposição em indivíduos idosos (65 a 85 anos de idade) é cerca de 13% mais elevada na AUC e cerca de 6% mais elevada na Cmax para o Apremilast do que nos indivíduos jovens (18 a 55 anos de idade). Os dados farmacocinéticos em indivíduos com mais de 75 anos de idade, em ensaios clínicos, são limitados. Não é necessário um ajuste posológico para os doentes idosos.
Compromisso renal
Não existe uma diferença significativa na farmacocinética do Apremilast em indivíduos com compromisso renal ligeiro ou moderado e indivíduos saudáveis emparelhados (n=8 em cada grupo). Os resultados sustentam não serem necessários ajustes de dose em doentes com compromisso renal ligeiro e moderado. A dose de Apremilast deve ser reduzida para 30 mg uma vez por dia em doentes com compromisso renal grave (TFGe inferior a 30 mL/min/1,73 m 2 ou CLcr < 30 mL/min). Em 8 indivíduos com compromisso renal grave aos quais foi administrada uma dose única de 30 mg de Apremilast, a AUC e a Cmax do Apremilast aumentaram em cerca de 89% e 42%, respetivamente.
Compromisso hepático
A farmacocinética do Apremilast e do seu principal metabolito M12 não é afetada por compromisso hepático moderado ou grave. Não é necessário um ajuste da dose em doentes com compromisso hepático.
Dados de segurança pré-clínica
Os dados não clínicos não revelam riscos especiais para o ser humano, segundo estudos convencionais de farmacologia de segurança e de toxicidade de dose repetida. Não existe evidência para potencial imunotóxico, de irritação dérmica ou fototóxico.
Fertilidade e desenvolvimento embrionário precoce
Num estudo de fertilidade com ratinhos machos, o Apremilast em doses orais de 1, 10, 25 e 50 mg/kg/dia não produziu efeitos na fertilidade masculina; o nível sem efeitos adversos observados (NOAEL) para a fertilidade masculina foi superior a 50 mg/kg/dia, 3 vezes superior à exposição clínica.
Num estudo combinado de fertilidade e toxicidade do desenvolvimento embriofetal em ratinhos fêmeas com doses orais de 10, 20, 40 e 80 mg/kg/dia, observou-se um prolongamento do ciclo estral e um aumento do tempo até ao acasalamento com doses de 20 mg/kg/dia e superiores; apesar disto, todos os ratinhos acasalaram e as taxas de gravidez não foram afetadas. O nível de efeito não observado (NOEL) para a fertilidade feminina foi de 10 mg/kg/dia (1,0 vez superior à exposição clínica).
Desenvolvimento embrio-fetal
Num estudo combinado de fertilidade e toxicidade do desenvolvimento embrio-fetal em ratinhos fêmeas com doses orais de 10, 20, 40 e 80 mg/kg/dia, os pesos absolutos e/ou relativos dos corações das mães aumentaram com doses de 20, 40 e 80 mg/kg/dia. Observou-se um aumento dos números de reabsorções precoces e diminuição do número de ossos társicos ossificados com 20, 40 e 80 mg/kg/dia. Observou-se uma redução dos pesos fetais e atraso na ossificação do osso supraoccipital do crânio com 40 e 80 mg/kg/dia. O NOEL materno e de desenvolvimento no ratinho foi de 10 mg/kg/dia (1,3 vezes superior à exposição clínica).
Num estudo de toxicidade do desenvolvimento embrio-fetal no macaco, doses orais de 20, 50, 200 e 1000 mg/kg/dia resultaram num aumento relacionado com a dose de perda pré-natal (abortos) com doses de 50 mg/kg/dia e superiores; não se observou qualquer efeito relacionado com o medicamento de ensaio nas perda pré-natais com 20 mg/kg/dia (1,4 vezes superior à exposição clínica).
Desenvolvimento pré e pós-natal
Num estudo pré e pós-natal, o Apremilast foi administrado por via oral a ratinhos fêmeas grávidas em doses de 10, 80 e 300 mg/kg/dia desde o dia 6 de gestação (DG) ao dia 20 de aleitamento. Observaram-se reduções e aumentos do peso corporal materno e uma morte associada a dificuldades no trabalho de parto com 300 mg/kg/dia. Foram também observados sinais físicos de toxicidade materna associada ao trabalho de parto num ratinho com 80 e 300 mg/kg/dia. Observou-se um aumento damorte das crias no período peri e pós-natal e uma redução dos pesos corporais das crias durante a primeira semana de aleitamento com doses ≥ 80 mg/kg/dia (≥ 4,0 vezes superior à exposição clínica).
Não se verificaram efeitos relacionados com o Apremilast na duração da gravidez, no número de ratinhas grávidas no fim do período de gestação, no número de ratinhas que deram à luzou quaisquer efeitos de desenvolvimento nas crias depois do dia 7 do período pós-natal. É provável que os efeitos de desenvolvimento observados nas crias durante a primeira semana do período pósnatal estivessem relacionados com a toxicidade na cria relacionada com o Apremilast (diminuição do peso das crias e viabilidade) e/ou ausência de cuidados maternos (maior incidência de ausência de leite no estômago das crias). Todos os efeitos do desenvolvimento foram observados durante a primeira semana do período pós-natal; não foram observados efeitos relacionados com o Apremilast durante os restantes períodos pré e pós-desmame, incluindo os parâmetros de maturação sexual, comportamentais, de acasalamento, fertilidade e uterinos. O NOEL para a toxicidade materna e para a geração F1 no ratinho foi de 10 mg/kg/dia (1,3 vezes superior à AUC clínica).
Estudos de carcinogenicidade
Os estudos de carcinogenicidade em ratinhos e ratos não mostraram haver evidência de carcinogenicidade relacionada com o tratamento com Apremilast.
Estudos de genotoxicidade
O Apremilast não é genotóxico. O Apremilast não induziu mutações num ensaio de Ames ou aberrações cromossómicas em culturas de linfócitos de sangue periférico humano na presença ou ausência de ativação metabólica. O Apremilast não foi clastogénico num ensaio de micronúcleo in vivo em ratinhos com doses até 2000 mg/kg/dia.
Outros estudos
Não existe evidência para potencial imunotóxico, de irritação dérmica ou fototóxico.