 Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)
Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)Qual a ação da substância do Lescol XL?
Resultados da Eficácia
Estudos clínicos
Em três estudos multicêntricos, duplo-cegos, ativo-controlados, em cerca de 1.700 pacientes com hipercolesterolemia primária ou dislipidemia mista, Fluvastatina Sódica 80 mg foi comparado a Fluvastatina Sódica 40 mg administrado na hora de dormir ou duas vezes ao dia, por mais de 24 semanas de tratamento.
As taxas de respostas no tempo em que a resposta terapêutica máxima é atingida são ilustradas na Figura 1 para doses de Fluvastatina Sódica 40 mg (média da redução de LDL-C de 26%) e doses de Fluvastatina Sódica 80 mg (média da redução de LDL-C de 36%).
Figura 1 - Taxa de resposta por faixa de percentual de redução de LDL-C na semana 4. (Resultados são agrupados a partir de 3 doses mais elevadas nos estudos comparativos).
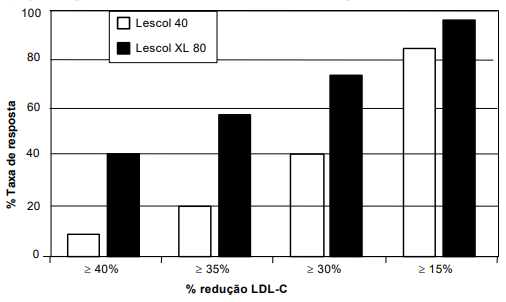
Nestes estudos, Fluvastatina Sódica 40 mg/Fluvastatina Sódica 80 mg reduziu significativamente o total-C, LDL-C, apo-B e TG, e aumentou o HDL-C após 24 semanas de tratamento de dose prescrita (vide Tabela 1).
Tabela 1- Porcentagem média de mudança em relação ao basal após 24 semanas (todos pacientes):
| Dose | Total-C | LDL-C | HDL-C | HDL-C (basal ≤ 35 mg/dL) | Apo-B | TG* |
| Fluvastatina Sódica 40 | -17% | -25% | +6% | +10% | -18% | -12% |
| Fluvastatina Sódica 80 | -23% | -34% | +9% | +14% | -26% | -19% |
*Porcentagem média de mudança.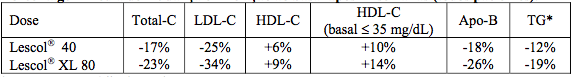
Dos 857 pacientes randomizados para Fluvastatina Sódica 80 mg, 271 com dislipidemia primária mista (Fredrickson Tipo IIb) definida por nível plasmático basal de triglicérides ≥ 200 mg/dL tiveram uma redução mediana no triglicérides de 25%. Nestes pacientes, Fluvastatina Sódica 80 mg produziu um aumento significativo no HDL-C de 13%. Este efeito foi ainda mais pronunciado naqueles pacientes com níveis basais de HDL-C muito baixos (por ex.: < 35 mg/dL), que tiveram um aumento médio no HDL-C de 16%. Também foi atingido um decréscimo significativo no total-C, LDL-C e apo-B (vide Tabela 2). Nestes estudos, foram excluídos pacientes com triglicérides > 400 mg/dL.
Tabela 2 - Porcentagem média de mudança em relação ao basal após 24 semanas (Dislipidemia primária mista):
| Dose | Total-C | LDL-C | HDL-C | Apo-B | TG* |
| Fluvastatina Sódica 40 | -17% | -23% | +7% | -17% | -18% |
| Fluvastatina Sódica 80 | -24% | -33% | +13% | -24% | -25% |
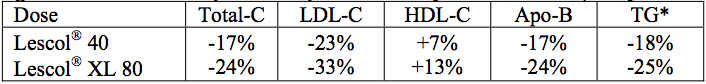 *Porcentagem média de mudança
*Porcentagem média de mudança
No Estudo de Aterosclerose Coronariana e Lipoproteína (LCAS), o efeito da fluvastatina na aterosclerose coronariana foi avaliado pela angiografia coronariana quantitativa em pacientes:
- Homens e mulheres (35-75 anos) com doença arterial coronariana e hipercolesterolemia leve a moderada (LDL-C basal 115-190 mg/dL). Neste estudo clínico controlado, randomizado, duplo-cego, 429 pacientes foram tratados com fluvastatina 40 mg/dia ou placebo. Os angiogramas coronarianos quantitativos foram avaliados no estado basal e após 2,5 anos de tratamento.
O tratamento com fluvastatina retardou a progressão das lesões ateroscleróticas coronarianas em 0,07 mm (intervalo de confiança de 95% para diferença entre os tratamentos de -0,1222 a -0,022 mm) após 2,5 anos, conforme medido pela alteração no diâmetro mínimo do lúmen (fluvastatina -0,028 mm vs. placebo -0,100 mm).
No Estudo de Prevenção com Intervenção de Fluvastatina Sódica (LIPS), o efeito da fluvastatina nos eventos cardíacos maiores (ECM) foi avaliado em pacientes:
- Homens e mulheres (18 a 80 anos) com doença coronariana cardíaca e uma ampla variação dos níveis de colesterol (estado basal CT: 3,5-7,0 mmol/L). Neste estudo randomizado, duplo-cego e placebo-controlado, administrou-se a fluvastatina (N = 844) 80 mg por dia por mais de 4 anos. O risco do primeiro evento cardíaco maior foi significativamente reduzido em 22% (p = 0,013) quando comparado ao placebo (N = 833). Estes efeitos benéficos foram particularmente notáveis em pacientes diabéticos e em pacientes com doenças multivasculares. A terapia com fluvastatina reduziu o risco de morte cardíaca e/ou infarto do miocárdio em 31% (p = 0,065).
Pacientes pediátricos
Em dois estudos abertos de dose titulada (ZA01 e 2301), a eficácia e a segurança da fluvastatina 20 a 80 mg foram investigadas durante um período de 2 anos para cada estudo, em um total de 113 crianças e adolescentes com hipercolesterolemia familiar heterozigótica.
Os estudos incluíram pacientes de 9 anos ou mais com um diagnóstico estabelecido de hipercolesterolemia familiar heterozigótica definido por:
- Níveis de LDL-C > 190 mg/dL (4,9 mmol/L);
- Ou níveis de LDL-C ≥ 160 mg/dL (4,1 mmol/L) e um ou mais fatores de risco [histórico familiar de doenças cardíacas coronarianas prematuras (CHD), fumo, hipertensão, colesterol de lipoproteína de alta densidade (HDL-C) < 35 mg/dL confirmado, diabetes mellitus];
- Ou defeito comprovado no ácido desoxirribonucleico (DNA) do receptor de LDL-C e níveis de LDL-C > 160 mg/dL (4,1 mmol/L) e níveis plasmáticos de triglicérides ≤ 600 mg/dL.
Os principais critérios de exclusão foram pacientes com hipercolesterolemia familiar homozigótica; formas secundárias de dislipoproteinemia; níveis plasmáticos de triglicérides > 600 mg/dL; ALT (TGP), AST (TGO) ou níveis de creatinina > 1,5 x LSN; CK plasmática ou TSH plasmático > 2 x LSN; IMC > 30 kg/m2 .
A dose inicial de fluvastatina foi 20 mg para a primeira semana e titulada (em um intervalo de 6 semanas) para 40 mg e então para 80 mg (duas vezes cápsulas de 40 mg ou comprimidos de liberação prolongada de 80 mg) se os níveis de LDL-C fossem > 3,2 mmol/L ou 3,4 mmol/L respectivamente.
A fluvastatina reduziu significativamente os níveis plasmáticos de total-C, LDL-C, TG, apo-B e aumentou HDL-C durante 2 anos de manutenção (vide Tabela 3).
Tabela 3 - Efeito da fluvastatina na redução de lípides em crianças e adolescentes com hipercolesterolemia familiar heterozigótica:
| Estudo ZA01 (pré-púbere) | Estado basal (mmol/L) N = 29 | 24 meses (mmol/L) N = 27 | % média de variação em relação ao estado basal (95% IC) N = 27 |
| Colesterol-LDL [média (DP)] | 5,8 (1,4) | 4,2 (1,5) | -27,0% (-34,7%, -19,4%) |
| Colesterol total [média (DP)] | 7,7 (1,4) | 5,9 (1,5) | -21,1% (-26,8%, -15,4%) |
| Colesterol-HDL [média (DP)] | 1,4 (0,3) | 1,4 (0,4) | 1,3% (-8,0%, 10,7%) |
| Triglicérides [mediana (faixa)] | 0,8(0,4-2,5) | 0,7(0,4-2,8) | -7,0% (-22,1%, 8,0%) |
| Estudo 2301 (pré-púbere, puberal e pós-púbere) | Estado basal (mmol/L) N = 84 | 24 meses (mmol/L) N = 84 | % média de variação em relação ao estado basal (95% IC) N = 84 |
| Colesterol-LDL [média (DP)] | 6,0 (1,27) | 4,1 (1,14) | -30,5% (-34,8%, -26,2%) |
| Colesterol total [média (DP)] | 7,7 (1,33) | 5,8 (1,16) | -23,6% (-27,2%, -19,9%) |
| Colesterol-HDL [média (DP)] | 1,2 (0,23) | 1,3 (0,23) | 5,0% (1,6%, 8,5%) |
| Triglicérides [mediana (faixa)] | 0,93(0,5-3,0) | 0,84(0,4-2,4) | -5,2% (-13,2%, 2,7%) |
Em ambos os estudos, todos os pacientes continuaram com seus crescimentos e maturação sexual normais. A fluvastatina não foi investigada em crianças abaixo de 9 anos.
Estes estudos não permitiram a extrapolação dos desfechos cardiovasculares com o início precoce da terapia com estatinas em crianças.
Referências bibliográficas:
1. A 28-week, double-blind and observer-blind to lipid values, randomized, parallel-group, multicenter, active-controlled study to assess the efficacy and safety of fluvastatin 80 mg slow release formulation administered at bedtime compared to fluvastatin (Lescol) 40 mg immediate release formulation administered at bedtime or twice daily in patients with primary hypercholesterolemia. Novartis Pharma AG, Basel, 10-Sep-99 [Study No. F302-E-00]. IRD – Lescol XL 80 Part IV A Volume 11, p. 001.
2. A 28-week, double-blind, and observer-blind to lipid values, active-controlled randomized, parallel-group, multicenter study to assess the safety and efficacy of fluvastatin 80 mg slow release form administered once daily at bedtime in patients with primary hypercholesterolemia compared to Lescol 40 mg. NPC, East Hanover, 30-Jul-99 [Study F351-E-00]. IRD – Lescol XL 80 Part IV A Volume 12, p. 001.
3. A 28-week, double-blind and observer-blind to lipid values, randomized, parallel-group, multicenter, positive controlled study to assess the efficacy and safety of fluvastatin 80 mg slow release formulation compared to fluvastatin (Lescol) 40 mg immediate release formulation (both administered at bedtime), and to fluvastatin (Lescol) 40 mg administered twice daily (BID) in patients with primary hypercholesterolemia. NPC, East Hanover, 30-Jul-99 [Study No. F353-E-00]. IRD – Lescol XL 80 Part IV A Volume 13, p. 001.
4. Lescol XL (fluvastatin slow release form) XUO 320 prolonged release tablets, 80 mg. Clinical Data Summary. 24- Nov-99. IRD – Lescol XL 80 Part IC.
5. Lescol XL (fluvastatin slow release form) XUO 320 prolonged release tablets, 80 mg. Expert Report on clinical documentation. 02-Nov-99. IRD – Lescol XL 80 Part IC.
6. Gotto AM. ER on clinical documentation. Indication: Coronary atherosclerosis in primary hypercholesterolemia 18- Dec-96 [Doc. # 162752.01 link 701-411].
7. Clinical Expert Report on secondary prevention of major adverse cardiac events. Novartis Pharmaceuticals Corporation East Hanover, New Jersey. 11 Jul 02. IRD – LIPS.
8. International, multicenter,randomized, double blind, placebo controlled study of the long term effects of fluvastatin (Lescol) on major adverse cardiac events in patients with coronary heart disease after successful first TransCatheter Therapy (TCT); Clinical Study Report LES-EUR-01; Novartis Pharma AG. Basle, Switzerland. 31 May 02. IRD – LIPS.
9. Clinical Overview on Fluvastatin pediatric indication in children with heterozygous familial hypercholesterolemia. Novartis Pharma AG. Basel, Switzerland. 31 Aug. 05.
10. Clinical Study Report on a prospective dose titration study of the efficacy and safety of Lescol (Fluvastatin) in the treatment of children with heterozygous familial hypercholesterolemia. Novartis Pharma AG. Basel, Switzerland. 03 Aug. 05.
11. Clinical Study Report on an open label, phase III, dose titration, multicenter study to assess the efficacy and safety of fluvastatin capsules and fluvastatin extended release (XL) tablets (20, 40 and 80 mg) given orally at bedtime for 114 weeks in pediatric patients with heterozygous familial hypercholesterolemia. Novartis Pharma AG. Basel, Switzerland. 25 Jul 05.
Características Farmacológicas
Grupo farmacoterapêutico: inibidores da HMG-CoA redutase.
Código ATC: C10A A04.
Farmacodinâmica
A fluvastatina, agente redutor de colesterol totalmente sintético, é inibidora competitiva da HMG-CoA redutase, a qual é responsável pela conversão da HMG-CoA em mevalonato, um precursor de esteróis, inclusive do colesterol. A fluvastatina exerce seu efeito principal no fígado e é essencialmente um composto racêmico de dois eritro-enantiômeros, sendo que um deles exerce a atividade farmacológica. A inibição da biossíntese do colesterol reduz o colesterol nas células hepáticas, o que estimula a síntese dos receptores das lipoproteínas de baixa densidade (LDL) e, portanto, aumenta a captação das partículas de LDL. O resultado definitivo desses mecanismos é a redução da concentração plasmática de colesterol.
Fluvastatina Sódica reduz o colesterol total (total-C), o colesterol LDL (LDL-C), a apolipoproteína B (apo-B) e os triglicérides (TG); e aumenta o colesterol HDL (HDL-C) em pacientes com hipercolesterolemia e dislipidemia mista. A resposta terapêutica é bem estabelecida dentro de 2 semanas, e a resposta máxima é atingida dentro de 4 semanas desde o início do tratamento e mantida durante o tratamento crônico.
Farmacocinética
Absorção
A fluvastatina é absorvida rápida e completamente (98%) após administração oral de uma solução em voluntários em jejum. Após a administração oral de Fluvastatina Sódica 80 mg, e em comparação com as cápsulas, a taxa de absorção da fluvastatina é quase 60% mais lenta, enquanto o tempo médio da permanência da fluvastatina é aumentado em aproximadamente 4 horas. Em indivíduos alimentados, o fármaco é absorvido em velocidade reduzida.
Distribuição
A fluvastatina exerce seu principal efeito no fígado, que é também o órgão principal para o seu metabolismo. A biodisponibilidade absoluta calculada a partir das concentrações sanguíneas sistêmicas é de 24%. O volume de distribuição aparente (Vz/f) para o fármaco é de 330 litros. Mais de 98% do fármaco circulante está ligado a proteínas plasmáticas e essa ligação não é afetada pela concentração de fluvastatina nem pela varfarina, ácido salicílico ou glibenclamida.
Metabolismo
A fluvastatina é principalmente metabolizada no fígado. Os principais componentes circulantes no sangue são a fluvastatina e o metabólito farmacologicamente inativo ácido N-desisopropil-propiônico. Os metabólitos hidroxilados têm atividade farmacológica, mas não apresentam circulação sistêmica. As vias do metabolismo hepático da fluvastatina em humanos têm sido completamente elucidadas. Existem várias vias alternativas à via do citocromo P450 (CYP450) para biotransformação da fluvastatina e dessa forma o metabolismo da fluvastatina é relativamente insensível à inibição do CYP450, a principal causa das interações medicamentosas.
Vários estudos in vitro detalhados reportaram o potencial inibitório da fluvastatina nas isoenzimas CYP comuns. A fluvastatina inibiu apenas o metabolismo de compostos que são metabolizados pelo CYP2C9. Apesar do potencial que existe para interação competitiva entre fluvastatina e compostos que são substratos do CYP2C9, como diclofenaco, fenitoína, tolbutamida e varfarina, os dados clínicos indicaram que este evento é improvável.
Eliminação
Após administração da fluvastatina-H3 em voluntários sadios, a excreção da radioatividade é de cerca de 6% na urina e 93% nas fezes e a fluvastatina responde por menos de 2% da radioatividade total excretada. O “clearance” (depuração plasmática) (CL/f) da fluvastatina no homem é calculado como sendo de 1,8 ± 0,8 L/min. Concentrações plasmáticas em “steady-state” (estado de equilíbrio) não indicam acúmulo de fluvastatina após administração de 80 mg diariamente. Após a administração oral de 40 mg de Fluvastatina Sódica, a meia-vida de distribuição terminal para a fluvastatina é de 2,3 ± 0,9 horas.
Não foram observadas diferenças significativas na ASC (área sob a curva) quando a fluvastatina foi administrada com a refeição noturna ou 4 horas após a mesma.
Populações especiais
Idade e gênero
As concentrações plasmáticas de fluvastatina não variam em função de idade ou sexo da população geral. Entretanto, foi observado um aumento da resposta ao tratamento nas mulheres e idosos.
Insuficiência hepática
Como a fluvastatina é eliminada principalmente pela via biliar e está sujeita a metabolismo pré-sistêmico significativo, existe potencial para o acúmulo do fármaco em pacientes com insuficiência hepática.
Insuficiência renal
A fluvastatina é depurada pelo fígado, com menos de 6% da dose administrada excretada na urina. A farmacocinética da fluvastatina permanece inalterada em pacientes com insuficiência renal leve a grave.
Dados de segurança pré-clínicos
Toxicidade aguda
O valor aproximado de DL50 de fluvastatina, administrada por via oral, é maior que 2 g/kg em camundongos e maior que 0,7 g/kg em ratos.
Toxicidade de dose repetida
A segurança da fluvastatina foi extensivamente investigada em estudos de toxicidade em ratos, coelhos, cães, macacos, camundongos e hamsters. Identificou-se uma variedade de alterações que são comuns aos inibidores da HMG-CoA redutase, por exemplo: hiperplasia e hiperqueratose de estômago não glandular de roedores; catarata em cães; miopatia em roedores; alterações hepáticas leves na maioria dos animais de laboratório, alterações na vesícula biliar em cães, macacos e hamsters, aumento de peso da tireoide em ratos; e degeneração testicular em hamsters. A fluvastatina não está relacionada a alterações degenerativas e vasculares do sistema nervoso central relatadas em cães que utilizaram outros membros desta classe de compostos.
Carcinogenicidade
Um estudo de carcinogenicidade foi realizado em ratos, utilizando-se dosagens de 6, 9 e 18 mg/kg por dia (atingindo a dose de 24 mg/kg por dia após 1 ano) para estabelecer uma dose máxima precisa de tolerabilidade. Estes níveis de dosagens levaram a níveis de concentração plasmática de aproximadamente 9, 13 e 26 a 35 vezes a concentração plasmática média do fármaco em humanos após uma dose oral de 40 mg. Na dose de 24 mg/kg por dia observou-se uma baixa incidência de papiloma de células escamosas no antro gástrico e um carcinoma na mesma região. Além disso, relatou-se um aumento da incidência de adenomas e carcinomas das células foliculares da tireoide em ratos machos tratados com 18 a 24 mg/kg por dia.
Um estudo de carcinogenicidade conduzido em camundongos sob níveis de dosagem equivalentes a 0,3; 15 e 30 mg/kg por dia revelou, assim como no estudo em ratos, um aumento estatisticamente significativo dos papilomas das células escamosas do antro gástrico em machos e em fêmeas sob doses de 30 mg/kg por dia e, em fêmeas, sob doses de 15 mg/kg por dia.
Estas dosagens produziram níveis de concentração plasmática aproximadamente 0,2; 10 e 21 vezes a concentração plasmática média em humanos após uma dose oral de 40 mg.
O estudo de carcinogenicidade em camundongos foi repetido em doses orais de 50, 150 e 350 mg/kg/dia. Não houve evidência de aumento de neoplasia nessas doses.
As neoplasias do antro gástrico observadas em ratos e camundongos refletem uma hiperplasia crônica causada, pela exposição e contato direto da fluvastatina do que por um efeito genotóxico do fármaco. O aumento da incidência de neoplasias das células foliculares da tireoide em ratos machos sob tratamento com fluvastatina parece ser consistente com achados espécie-específicos com outros inibidores de HMG-CoA redutase. Contrariamente a estes outros inibidores, não há relatos de aumentos na incidência de adenomas ou carcinomas hepáticos relacionados ao tratamento.
Mutagenicidade
Não se observou nenhuma evidência de mutagenicidade in vitro, com ou sem ativação do metabolismo hepático em ratos, nos seguintes estudos: testes mutagênicos microbianos usando cepas mutantes de Salmonella typhimurium ou Escherichia coli; ensaio de transformação maligna em células de BALB/3T3; síntese não programada de DNA em hepatócitos primários de ratos; aberrações cromossômicas em células de hamsters chineses V79; células de hamsters chineses HGPRT V79. E também, não houve evidência de mutagenicidade in vivo tanto em testes de micronúcleos de ratos quanto de camundongos.
Toxicidade reprodutiva
Em um estudo realizado em ratos com doses de 0,6; 2 e 6 mg/kg por dia, administradas em fêmeas e doses de 2; 10 e 20 mg/kg por dia, administradas em machos, a fluvastatina não apresentou reações adversas na fertilidade ou no desempenho da reprodução. Os estudos de teratologia em ratos (1, 12 e 36 mg/kg) e em coelhos (0,05; 1 e 10 mg/kg) revelaram toxicidade materna sob níveis de altas doses, mas não houve evidência de potencial teratogênico ou embriotóxico. Um estudo em ratas recebendo doses de 12 e 24 mg/kg por dia, durante o período final de gestação até o desmame dos filhotes, resultou em mortalidade materna no final da gravidez ou próximo a este período e no pós-parto, bem como em letalidade fetal e neonatal. Não ocorreram efeitos nas fêmeas grávidas ou nos fetos sob a baixa dosagem de 2 mg/kg por dia.
Um segundo estudo com doses de 2; 6; 12 e 24 mg/kg por dia, durante o término da gestação e o início da lactação, revelou efeitos similares aos causados por cardiotoxicidade, sob administração de doses de 6 mg/kg por dia ou acima deste valor. Em um terceiro estudo, as ratas grávidas receberam doses de 12 ou 24 mg/kg por dia, durante o final da gestação até o desmame dos filhotes, com ou sem a suplementação concomitante de ácido mevalônico, um derivado da HMG-CoA que é essencial para a biossíntese do colesterol. A administração concomitante do ácido mevalônico preveniu completamente a cardiotoxicidade e a mortalidade materna e neonatal. Portanto, a letalidade materna e neonatal observada com a fluvastatina reflete seu pronunciado efeito farmacológico durante a gravidez.