 Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)
Revisado por Isabelle Baião de Mello Neto (CRF-MG 24309)Qual a ação da substância do Koselugo?
Resultados de Eficácia
Eficácia clínica
Sprint
A eficácia de Sulfato de Selumetinibe foi avaliada em um estudo aberto, multicêntrico, de braço único [Estrato 1 do SPRINT Fase II (NCT01362803)] de 50 pacientes pediátricos com NF1 e NP inoperável que causava morbidade significativa. NP inoperável foi definido como um NP que não poderia ser completamente removido cirurgicamente sem risco de morbidade substancial devido ao revestimento, ou proximidade, de estruturas vitais, grau de invasão ou alta vascularização do NP. Pacientes receberam 25 mg/m2 (ASC) duas vezes ao dia, por 28 dias (1 ciclo de tratamento) em um esquema posológico contínuo. O tratamento foi descontinuado se o paciente não estivesse mais obtendo benefício clínico, apresentasse toxicidade inaceitável ou progressão do NP ou a critério do investigador.
O NP alvo, o NP que causou sintomas clínicos ou complicações relevantes (morbidades relacionadas com NP) foi avaliado para taxa de resposta usando análise de ressonância magnética nuclear (RMN) volumétrica de leitura centralizada de acordo com os critérios de Avaliação de Resposta em Neurofibromatose e Schwannomatose (REiNS). A resposta do tumor foi avaliada no basal e durante o tratamento a cada 4 ciclos por 2 anos e então a cada 6 ciclos.
Os pacientes tiveram avaliações de RMN volumétrica no NP alvo, avaliações de resultado clínico, avaliação funcional e relataram resultados pertinentes aos sintomas clínicos, avaliação da dor e qualidade de vida relacionada à saúde.
A mediana de idade dos pacientes foi de 10,2 anos (intervalo: 3,5 – 17,4 anos), 60% eram homens, 84% eram caucasianos.
As características da doença no basal são fornecidas na Tabela 1.
Tabela 1 Características Basais da Doença
| Características | SPRINT (N = 50) |
| Volume NP alvo (mL) | |
| Mediana (intervalo) | 487,5 (5,6 - 3820) |
| Número de morbidades relacionadas ao NP | |
| Mediana (intervalo) | 3 (1 - 4) |
| Morbidades relacionadas ao NP alvo (%) | |
| Desfiguração | 88% |
| Disfunção motora | 66% |
| Dor | 52% |
| Disfunção das vias aéreas | 32% |
| Comprometimento visual | 20% |
| Disfunção na bexiga/intestinal | 20% |
O desfecho primário de eficácia foi Taxa de Resposta Objetiva (TRO), definida como o percentual de pacientes com resposta completa (definida como desaparecimento do NP alvo) ou resposta parcial confirmada (definida como redução ≥20% no volume do NP, confirmada por uma avaliação tumoral subsequente dentro de 3-6 meses), com base na avaliação centralizada do NCI (National Cancer Insitute). A Duração da Resposta (DR) também foi avaliada.
O desfecho primário, TRO foi de 66% (IC 95%, 51,2 – 78,8). O tempo para início da resposta para a maioria dos pacientes (24/33 [72,7%]) ocorreu dentro de 8 ciclos (intervalo 4 – 20 ciclos).
A mediana da DR desde o início da resposta não foi atingida; no momento do corte de dados, a mediana do tempo de acompanhamento foi de 24 ciclos. Dos 33 pacientes que tiveram respostas parciais confirmadas, 29 (87,9%) permaneceram em resposta após 12 ciclos; 4 pacientes foram censurados não devido à progressão. A probabilidade de permanecer em resposta após 12 e 16 ciclos, estimada usando o método de Kaplan-Meier, foi de 100% (IC 95% não estimado) e 96,2% (IC 95% 75,7 – 99,4), respectivamente. A mediana do tempo do início do tratamento até progressão da doença durante o tratamento não foi atingida.
Tabela 2 Resultados de eficácia NF1 NP do SPRINT
| Parâmetro de Eficácia | SPRINT (N = 50) |
| Taxa de Resposta Objetiva a | |
| Taxa de Resposta Objetiva, % (IC 95%) | 66,0 (51,2 – 78,8) |
| Melhor resposta objetiva, n (%) b, c | |
| Resposta Completa | 0 |
| Resposta Parcial Confirmada | 33 (66%) |
| Resposta Parcial Não Confirmada | 4 (8%) |
| Doença Estável | 11 (22%) |
| Doença Progressiva | 0 |
| Duração da Resposta d | |
| Mediana (IC 95%) meses | NA (NE – NE) |
IC – intervalo de confiança, NE – não estimado, NA – não atingido.
a Respostas necessitaram de confirmação pelo menos 3 meses após os critérios para primeira resposta parcial terem sido preenchidos.
b Resposta completa: desaparecimento da lesão alvo; Resposta Parcial: redução no volume do NP alvo em ≥20% em comparação com o basal; Doença Estável: alteração insuficiente do volume do basal para qualificar resposta parcial ou doença progressiva; Doença Progressiva: aumento no volume do NP alvo em ≥20% em comparação com o basal ou o tempo de melhor resposta documentada.
c Dois pacientes não foram avaliáveis.
d Com base na análise de Kaplan-Meier em pacientes com resposta parcial confirmada.
No momento do corte de dados, 28 (56%) pacientes permaneciam em resposta parcial confirmada, 2 (4%) apresentavam resposta parcial não confirmada, 15 (30%) tinham doença estável e 3 (6%) tinham doença progressiva.
A mediana da melhor alteração percentual no volume de NP do basal foi -27,85% (intervalo: 2,2% a -54,5%). A Figura 1 demonstra a melhor alteração percentual no volume de NP alvo para cada paciente.
Figura 1 Gráfico tipo cascata da melhor alteração percentual do basal no volume de NP-alvoa
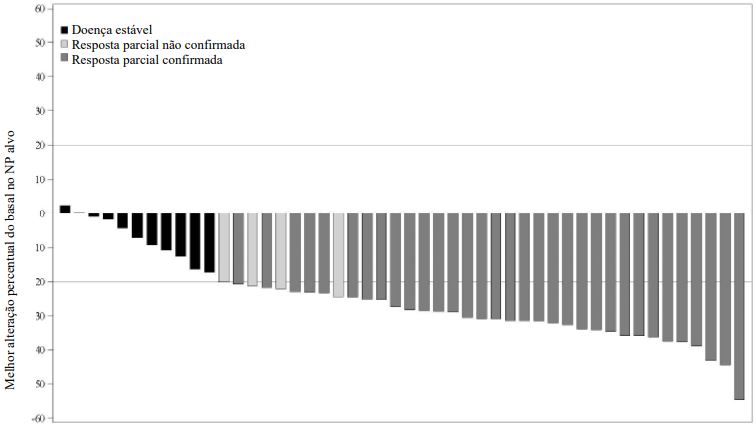
a Melhor alteração percentual no volume de NP alvo é a redução máxima do basal, ou aumento mínimo do basal na ausência de uma redução. Dois pacientes foram não avaliáveis.
Avaliações de Resultado Clínico
A intensidade da dor do NP alvo foi autorrelatada pelos pacientes ≥8 anos de idade usando uma Escala de Classificação Numérica de 11 pontos (NRS-11). Uma redução clinicamente significativa na dor (definida como uma redução ≥2 pontos do basal) foi reportada por 50% dos pacientes (n=12) no pré-Ciclo 13; 12 pacientes (50%) não reportaram alteração (10 dos quais tinham um escore basal ≤1) e nenhum paciente mostrou deterioração.
A qualidade de vida relacionada à saúde (QVRS) reportada pelos pais (todos os pacientes) e reportada pelo paciente (≥8 anos de idade) foi avaliada usando o questionário Peds-QL. Com base em uma análise MMRM, uma melhora clinicamente significativa na QVRS (limiar clinicamente significativo 11,9) foi reportada pelos pais no pré-Ciclo 13 com uma alteração média do basal no escore total do Peds-QL de 12,7 (IC 95% 8,91 – 16,55). A melhora na QVRS também foi reportada pelos pacientes com uma alteração média do basal de 6,68 (IC 95% 1,34 – 12,02).
Referências Bibliográficas
Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, et al. Activity of Selumetinib in Neurofibromatosis Type 1-Related Plexiform Neurofibromas. N Engl J Med. 2016;375(26):2550-2560.
Características Farmacológicas
Mecanismo de ação
Selumetinibe é um potente e seletivo inibidor das proteínas quinases ativadas por mitógenos, quinases 1 e 2 (MEK1/2), não competitivo com relação ao ATP e disponível por via oral. As proteínas MEK1/2 são componentes críticos da via RAF-MEK-ERK regulada por RAS, que é frequentemente ativada em diferentes tipos de tumores. Selumetinibe bloqueia a atividade da MEK e inibe o crescimento de linhagens celulares ativadas pela via RAF-MEK-ERK. Portanto, a inibição de MEK pode bloquear a proliferação e sobrevivência das células tumorais, nas quais a via RAF-MEK-ERK está ativada.
Propriedades Farmacodinâmicas
Em modelos de camundongos geneticamente modificados de NF1 que geram neurofibromas que recapitulam o genótipo e fenótipo de neurofibromas humanos do tipo 1, a administração oral de selumetinibe inibe a fosforilação de ERK, reduz o volume, proliferação, número e crescimento de neurofibroma.
Eletrofisiologia cardíaca
Em um estudo positivo (moxifloxacino) controlado por placebo, o efeito de selumetinibe no intervalo QTc, em 48 adultos sadios, após uma dose única oral de 75 mg, não demonstrou ser clinicamente relevante (alteração <10 ms). Uma análise farmacocinética-farmacodinâmica previu uma alteração <10 ms com a dose de 150 mg (3 vezes maior do que a dose máxima recomendada de 50 mg em pacientes pediátricos com NF1).
Propriedades Farmacocinéticas
Na posologia recomendada de 25 mg/m2 duas vezes ao dia em pacientes pediátricos (3 a ≤18 anos de idade), a média geométrica (coeficiente de variação [CV%]) da concentração plasmática máxima (Cmax) foi 731 (62%) ng/mL e a área sob a curva da concentração plasmática (AUC0-12) após a primeira dose foi 2009 (35%) ng·h/mL. Acúmulo mínimo de ~1,1 vezes foi observado no estado estacionário com a administração duas vezes ao dia.
Em pacientes pediátricos, com um nível de dose de 25 mg/m2, selumetinibe tem uma depuração oral aparente de 8,8 L/h, volume médio de distribuição no estado de estacionário de 78 L e uma meia-vida média de eliminação de ~6,2 horas.
Absorção
Em indivíduos adultos sadios, a média da biodisponibilidade absoluta oral de selumetinibe foi 62%. Após a administração oral, selumetinibe é rapidamente absorvido, produzindo concentrações plasmáticas máximas no estado de estacionário (Tmax) entre 1-1,5 horas após a dose.
Efeito do alimento
Em estudos clínicos separados, em indivíduos adultos sadios e em pacientes adultos com malignidades sólidas avançadas, a administração concomitante de 75 mg de selumetinibe com uma refeição com alto teor de gordura resultou em uma redução média na Cmax de 50% e 62%, respectivamente em comparação com a administração em jejum. A média da AUC de selumetinibe foi reduzida em 16% e 19%, respectivamente, e o tempo para atingir a concentração máxima (Tmax) foi retardado em aproximadamente 1,5 horas.
Em indivíduos adultos sadios, a administração concomitante de 50 mg de selumetinibe com uma refeição com baixo teor de gordura resultou em Cmax 60% menor em comparação com a administração em jejum. A AUC de selumetinibe foi reduzida em 38% e o tempo para atingir a concentração máxima (Tmax) foi retardado em aproximadamente 0,9 horas.
Distribuição
O volume médio aparente de distribuição de selumetinibe entre 20 a 30 mg/m2 no estado estacionário variou de 78 a 171 L em pacientes pediátricos, indicando distribuição moderada no tecido.
A ligação in vitro à proteína plasmática é de 98,4% em humanos. Selumetinibe se liga principalmente à albumina sérica (96,1%) do que à α-1 glicoproteína ácida (<35%).
Biotransformação / Metabolismo
In vitro, selumetinibe passa por reações metabólicas de Fase 1 incluindo oxidação da cadeia lateral, Ndesmetilação e perda da cadeia lateral para formar metabólitos amido e ácido. CYP3A4 é a isoforma predominante responsável pelo metabolismo oxidativo de selumetinibe com CYP2C19, CYP1A2, CYP2C9, CYP2E1 e CYP3A5 envolvidas em menor extensão. Os estudos in vitro indicam que selumetinibe também passa por reações metabólicas diretas de Fase 2 para formar conjugados glicuronídeos envolvendo principalmente as enzimas UGT1A1 e UGT1A3. Glicuronidação é uma via significativa de eliminação para os metabólitos de Fase 1 de selumetinibe envolvendo diversas isoformas de UGT. Com base nos estudos in vitro, estima-se que cerca de 56% da depuração intrínseca observada de selumetinibe poderia ser atribuída ao metabolismo CYP e cerca de 29% atribuída à glicuronidação direta pelas enzimas UGT (ou seja, excluindo as vias onde a oxidação de Fase 1 ocorre antes de glicuronidação).
O metabólito ativo de Fase 1, N-desmetil selumetinibe é gerado principalmente pelo CYP2C19 e metabolizado através das mesmas vias que o selumetinibe. N-desmetil selumetinibe representa menos que 10% dos níveis de selumetinibe no plasma humano, mas é aproximadamente 3 a 5 vezes mais potente que o composto original, contribuindo para cerca de 21% a 35% da atividade farmacológica geral.
Eliminação
Em voluntários adultos sadios, após uma dose única oral de 75 mg de selumetinibe radiomarcado, 59% foi recuperado nas fezes (19% inalterado) enquanto que 33% da dose administrada (<1% como original) foi encontrada na urina 9 dias após a coleta da amostra.
Interações com proteínas transportadoras
Estudos in vitro sugerem que selumetinibe não inibe a proteína de resistência ao câncer de mama (PRCM), glicoproteína P (gp-P), OATP1B1, OATP1B3, OCT2, OAT1, MATE1 e MATE2K na dose pediátrica recomendada. Um efeito clinicamente relevante na farmacocinética de substratos de OAT3 administrados concomitantemente não pode ser excluído.
Efeitos do transportador gp-P e PRCM no selumetinibe
Com base nos estudos in vitro, selumetinibe é um substrato para transportadores PRCM e gp-P, mas é improvável que resulte em interações medicamentosas clinicamente significativas na dose pediátrica recomendada.
Populações especiais
Insuficiência renal
A exposição de 50 mg de selumetinibe oral foi investigada em indivíduos adultos com função renal normal (n=11) e indivíduos com doença renal em estágio terminal (DRET) (n=12). O grupo DRET demonstrou Cmax e AUC 16% e 28% menores, respectivamente, com a fração de selumetinibe não ligado sendo 35% maior em indivíduos DRET. Como resultado, as razões de Cmax e AUC não ligado foram 0,97 e 1,13 no grupo DRET quando comparadas com o grupo com função renal normal. Um pequeno aumento, aproximadamente 20% da AUC, na razão do metabólito N-desmetil/original foi detectado no grupo DRET quando comparado com o grupo normal. Como a exposição em indivíduos DRET foi similar àquela com função renal normal, investigações em indivíduos com comprometimento renal leve, moderado e grave não foram realizadas. Não é esperado que a insuficiência renal tenha influência significativa na exposição de selumetinibe.
Insuficiência hepática
Indivíduos adultos com função hepática normal (n=8) e insuficiência hepática leve (Child-Pugh A, n=8) receberam uma dose de 50 mg de selumetinibe, indivíduos com insuficiência hepática moderada (Child-Pugh B, n=8) uma dose de 50 ou 25 mg e indivíduos com insuficiência renal grave (Child-Pugh C, n=8) uma dose de 20 mg. A AUC da dose total normalizada de selumetinibe e AUC da dose não ligada foi 86% e 69% respectivamente, nos pacientes com insuficiência hepática leve em comparação com os valores de AUC dos indivíduos com função hepática normal. A exposição ao selumetinibe (AUC) foi maior em pacientes com insuficiência hepática moderada (Child-Pugh B) e grave (Child-Pugh C); os valores de AUC total e AUC não ligada dos indivíduos com função hepática normal foram, respectivamente, 159% e 141% (Child-Pugh B) e 157% e 317% (Child-Pugh C).
Pacientes asiáticos
A exposição ao selumetinibe parece ser maior em voluntários adultos sadios japoneses, asiáticos não japoneses e indianos em comparação com voluntários adultos ocidentais. No entanto, há sobreposição considerável com indivíduos ocidentais quando corrigido para peso corporal ou ASC.
Pacientes adultos (>18 anos de idade)
Os parâmetros farmacocinéticos em voluntários sadios adultos e pacientes adultos com malignidades sólidas avançadas, são similares àqueles em pacientes pediátricos (3 a ≤18 anos de idade) com NF1.
Em pacientes adultos com malignidades sólidas, a uma dose de 75 mg duas vezes ao dia, Cmax e média geométrica da AUC (%CV) foram 1307 (76%) ng/mL e 4736 (37%) ng·h/mL, respectivamente. As concentrações plasmáticas máximas de selumetinibe foram atingidas 1,5 horas após a dose com uma meia-vida média de eliminação de 7,8 horas. Cmax e AUC aumentaram proporcionalmente ao longo de um intervalo de dose de 25 mg a 150 mg e a administração de 75 mg de selumetinibe duas vezes ao dia resultou em acúmulo mínimo de ~1,2 vezes.
Dados pré-clínicos de segurança
Mutagenicidade
Selumetinibe não demonstrou potencial mutagênico ou clastogênico in vitro, mas produziu um aumento nos eritrócitos imaturos micronucleados (aberrações cromossômicas) em estudos de micronúcleos em camundongo, predominantemente via um modo de ação aneugênico. A exposição média livre (Cmax) no Nível Sem Eventos Adversos Observados (NOEL) foi aproximadamente 27 vezes maior do que a exposição clínica livre na dose máxima recomendada para humanos (DMRH) de 25 mg/m2.
Carcinogênese
Selumetinibe não foi carcinogênico em um estudo de 6 meses em camundongos transgênicos rasH2 em exposições livres 24 vezes (fêmeas) e 16 vezes (machos) a AUC clínica livre no DMRH e em um estudo de carcinogenicidade de 2 anos em ratos em exposições livres 2,9 vezes (fêmeas) e 3,7 vezes (machos) a AUC clínica livre no DMRH.
Toxicidade de dose repetida
Nos estudos de toxicidade de dose repetida em camundongos e ratos, os principais efeitos observados após a exposição de selumetinibe foram na pele, crostas de feridas associadas com erosões microscópicas e ulceração em ratos em uma exposição livre similar à exposição clínica (AUC livre) no DMRH; achados inflamatórios e ulcerativos no trato GI de camundongos associados com alterações secundárias no fígado e sistema linforeticular nas exposições livres aproximadamente 28 vezes a exposição clínica livre no DMRH; displasia de placa de crescimento (fiseal) em ratos machos em uma exposição livre 11 vezes a exposição livre clínica no DMRH. Os achados GI demonstraram evidência de reversibilidade após um período de recuperação. A reversibilidade para toxicidades cutâneas e displasia fiseal não foram avaliadas.
Toxicologia Reprodutiva
Fertilidade
Em um estudo de 6 meses com camundongos, selumetinibe não afetou o desempenho de acasalamento de machos em qualquer dose até 20 mg/kg duas vezes ao dia correspondente a aproximadamente 22 vezes a exposição clínica humana com base no AUC livre no DMRH. Em camundongos fêmeas expostas a selumetinibe a 12,5 mg/kg duas vezes ao dia, o desempenho de acasalamento e fertilidade não foram afetados, mas o número de fetos vivos foi levemente reduzido. Após o período de descontinuação de tratamento de três semanas, nenhum efeito foi aparente em qualquer parâmetro. O nível sem evento adverso observável (NOEL) para a toxicidade materna e efeitos no desempenho reprodutivo foi 2,5 mg/kg duas vezes ao dia (aproximadamente, 3,5 vezes a exposição livre em humanos na DMRH).
Toxicidade embriofetal
Nos estudos de desenvolvimento embriofetal em camundongos, selumetinibe causou uma redução no número de fetos vivos devido a um aumento na perda pós-implantação, uma redução na média dos pesos fetais e da ninhada, ocorrência aumentada de olho aberto e fenda palatina em níveis de dose que não induziram toxicidade materna significativa. Estes efeitos foram observados a uma exposição >3,5 vezes a exposição clínica no DMRH com base no AUC livre e indicam que selumetinibe pode ter potencial para causar defeitos no feto.
Desenvolvimento pré e pós-natal
A administração de selumetinibe em camundongos prenhes a partir do Dia 6 da gestação até o Dia 20 da lactação resultou em pesos corporais reduzidos dos filhotes e menos filhotes dentro dos critérios de constrição de pupila no Dia 21 após o parto. A incidência de malformações (olhos prematuramente abertos e fenda palatina) foi aumentada em todos os níveis de dose. As malformações ocorreram em concentração materna (Cmax) 0,4 vezes abaixo da média de concentração clínica livre na DMRH.
Selumetinibe e seu metabólito ativo foram excretados no leite de camundongos amamentando em concentrações aproximadamente iguais às do plasma.