 Revisado clinicamente por: Isabelle Baião de Mello Neto (CRF-MG 24309). Atualizado em: 01 de outubro de 2025.
Revisado clinicamente por: Isabelle Baião de Mello Neto (CRF-MG 24309). Atualizado em: 01 de outubro de 2025.Amvuttra: Para que serve? Indicações
O componente principal do Amvuttra® é a vutrisirana sódica.
Amvuttra® é um remédio que trata uma doença de família chamada amiloidose hereditária ATTR (hATTR). Essa condição acontece por falhas numa proteína do corpo chamada 'transtirretina' (TTR). Essa proteína é feita principalmente no fígado e transporta vitamina A e outras substâncias pelo organismo.
Amvuttra® é indicado apenas para adultos.
Como funciona o Amvuttra?
Em quem tem amiloidose hATTR, pedaços pequenos da proteína TTR se juntam formando depósitos chamados "amiloides". Esses depósitos podem se acumular nos nervos, coração e outras partes do corpo, atrapalhando seu funcionamento normal. Isso causa os sintomas da doença.
Amvuttra® age diminuindo a quantidade de proteína TTR que o fígado produz. Assim, menos proteína TTR fica no sangue para formar amiloide. Isso ajuda a reduzir os efeitos da doença.
Contraindicações do Amvuttra
Não use Amvuttra® se:
Você já teve reação alérgica grave à vutrisirana ou qualquer componente deste remédio. Em caso de dúvida, consulte seu médico antes de usar.
Como usar o Amvuttra?
A dose certa de Amvuttra® é de 25 mg a cada 3 meses.
Amvuttra® é aplicado como injeção sob a pele (subcutânea) na barriga, parte de cima do braço ou coxa. Um médico, enfermeiro ou farmacêutico aplicará este remédio.
Seu médico dirá quanto tempo você precisa usar Amvuttra®. Não pare o tratamento sem orientação médica.
Siga as orientações do seu médico sobre horários, doses e tempo de tratamento. Não interrompa o tratamento sem consultá-lo.
O que fazer se esquecer de usar o Amvuttra?
Se perder uma aplicação de Amvuttra®, entre em contato com seu médico logo para remarcar.
Em caso de dúvidas, consulte seu médico ou farmacêutico.
Precauções ao usar o Amvuttra
Converse com seu médico antes de usar este remédio.
Níveis baixos de vitamina A e suplementos
Amvuttra® reduz a vitamina A no seu sangue. Seu médico receitará um suplemento diário de vitamina A.
Sintomas da falta de vitamina A podem incluir:
- Problemas de visão (especialmente à noite), olhos secos, visão embaçada.
Se notar mudanças na visão ou outros problemas oculares durante o uso de Amvuttra®, informe seu médico. Ele pode encaminhá-lo a um oftalmologista.
Crianças e adolescentes
Amvuttra® não é indicado para menores de 18 anos.
Dirigir e operar máquinas
É pouco provável que Amvuttra® atrapalhe dirigir ou operar máquinas. Seu médico avaliará se sua condição permite essas atividades.
Gravidez e prevenção
Antes de começar o tratamento com Amvuttra®, avise seu médico se está grávida, suspeita de gravidez ou planeja engravidar. Não use este remédio na gravidez, exceto sob orientação médica.
Amvuttra® reduz os níveis de vitamina A, importante para o desenvolvimento do bebê. Os níveis podem ficar baixos por mais de 12 meses após a última dose. Se puder engravidar, use métodos contraceptivos eficazes durante o tratamento. Converse com seu médico sobre as opções. A gravidez deve ser descartada antes de iniciar o tratamento. Se engravidar durante o uso, avise seu médico imediatamente.
Este remédio não deve ser usado por grávidas sem orientação médica.
Amamentação
Antes de iniciar o tratamento com Amvuttra®, informe se está amamentando. Não se sabe se o remédio passa para o leite. Seu médico avaliará os benefícios para você versus os riscos para o bebê.
Efeitos colaterais do Amvuttra
Como todo remédio, este medicamento pode causar efeitos colaterais, que não aparecem em todas as pessoas.
Podem ocorrer os seguintes efeitos com Amvuttra®:
Muito Comum: pode afetar mais de 1 em cada 10 pessoas
- Dor nas juntas (artralgia);
- Dor nos braços e pernas.
Comum: pode afetar até 1 em cada 10 pessoas
- Dificuldade para respirar (falta de ar);
- Vermelhidão, dor, coceira, hematoma ou calor no local da injeção.
Atenção: este é um remédio novo. Embora pesquisas mostrem segurança e eficácia, podem ocorrer efeitos desconhecidos. Se isso acontecer, informe seu médico.
Apresentações do Amvuttra
Solução injetável 25 mg/0,5 mL
Vem em cartucho com 1 seringa pronta com 0,5 mL de solução (25 mg de vutrisirana).
Cada mL da solução contém 50 mg de vutrisirana (equivalente a 53 mg de vutrisirana sódica).
Uso subcutâneo.
Para adultos.
Composição do Amvuttra
Cada mL de solução contém:
53 mg de vutrisirana sódica (equivalente a 50 mg de vutrisirana).
Componentes adicionais: fosfato de sódio monobásico di-hidratado, fosfato de sódio dibásico di-hidratado, cloreto de sódio, água para injetáveis, hidróxido de sódio (ajuste de pH) e ácido fosfórico (ajuste de pH).
Superdose do Amvuttra: o que fazer?
Este remédio será aplicado por profissional de saúde. Se receber dose maior que a recomendada, seu médico verificará possíveis efeitos colaterais.
Em caso de superdose, busque socorro médico imediatamente e leve a embalagem ou bula. Ligue para 0800 722 6001 para orientações.
Interações com outros remédios
Informe seu médico sobre todos os remédios que usa, incluindo os sem receita.
Avise seu médico sobre qualquer outro medicamento em uso.
Não use remédios sem conhecimento do seu médico. Pode ser perigoso.
Interações com alimentos
Não aplicável.
Como o Amvuttra age no corpo?
Resultados de Eficácia
Classe Terapêutica ou Farmacológica
Classe farmacológica: pequeno ácido ribonucleico de interferência (siRNA).
Grupo farmacoterapêutico/código ATC: Outros medicamentos do sistema nervoso/N07XX18.
Eficácia
Vutrisirana Sódica contém vutrisirana, um pequeno ácido ribonucleico de interferência de cadeia dupla (siRNA) quimicamente modificado, que atua especificamente sobre o RNA mensageiro (mRNA) da transtirretina (TTR) mutante e do tipo selvagem, e está covalentemente ligado a um ligante contendo três resíduos de N-acetilgalactosamina (GalNAc), que permite a entrega do siRNA aos hepatócitos. O oligonucleotídeo quimicamente modificado aumentou a estabilidade metabólica, prolongou o tempo de residência no fígado e permite dosagem infrequente.
A fórmula estrutural do princípio ativo vutrisirana sódica é apresentada abaixo:
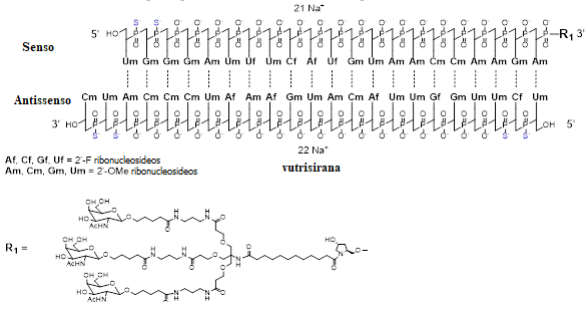
“O” denota ligação fosfodiester.
“S” denota ligação fosforotiato.
Linhas tracejadas denotam emparelhamento de bases Watson-Crick.
A fórmula molecular da vutrisirana (ácido livre) é C530H715F9N171O323P43S6 com peso molecular de 16.345 Da.
A eficácia de Vutrisirana Sódica foi demonstrada em um estudo clínico global, randomizado e aberto (HELIOS-A) em adultos com amiloidose hATTR com polineuropatia. Os pacientes foram distribuídos em 3:1 para receber 25 mg de Vutrisirana Sódica (N=122) via subcutânea, uma vez a cada 3 meses, ou 0,3 mg/kg de patisirana (N=42) por via intravenosa a cada 3 semanas, como grupo de referência.
O período de tratamento durou 18 meses, com análises no Mês 9 e Mês 18. Noventa e sete por cento (97%) dos pacientes tratados com Vutrisirana Sódica completaram pelo menos 18 meses do tratamento. As avaliações de eficácia compararam o grupo de vutrisirana com um grupo placebo externo (grupo placebo do estudo APOLLO Fase 3) com pacientes semelhantes. A avaliação da não inferioridade da redução da TTR sérica baseou-se na comparação entre o grupo de vutrisirana e o grupo de referência de patisirana dentro do estudo.
Entre os pacientes que receberam Vutrisirana Sódica, a idade mediana foi de 60 anos e 65% eram homens. Setenta por cento (70%) eram caucasianos, 17% asiáticos, 3% negros e 9% outros. Os pacientes eram da Europa Ocidental (35%), América do Norte (22%) ou outras regiões (43%). Foram observadas 22 variantes de TTR: V30M (44%), T60A (13%), E89Q (8%), A97S (6%), S50R (4%), V122I (3%), L58H (3%) e outras (18%). Vinte por cento (20%) tinham genótipo V30M com início precoce de sintomas (<50 anos). No início, 69% estavam no estágio 1 (caminhada sem dificuldade; neuropatia leve) e 31% no estágio 2 (necessidade de apoio para caminhar; comprometimento moderado). Sessenta e um por cento (61%) já haviam usado estabilizadores de TTR.
Segundo a classificação da New York Heart Association (NYHA) para insuficiência cardíaca, 9% eram classe I e 35% classe II. Trinta e três por cento (33%) atendiam critérios para envolvimento cardíaco (espessura da parede do ventrículo esquerdo ≥13 mm sem histórico de hipertensão ou doença da válvula aórtica).
Avaliações no Mês 9
O principal resultado foi a mudança no escore modificado de comprometimento da neuropatia +7 (mNIS+7) do início até o Mês 9. Esta medida avalia neuropatia motora, sensorial e autonômica, variando de 0 a 304 pontos, onde maior pontuação indica piora.
O tratamento com Vutrisirana Sódica melhorou a neuropatia em 17 pontos no mNIS+7 versus placebo (p<0,0001), com mudança média de -2,2 pontos (melhora) com Vutrisirana Sódica, versus aumento de 14,8 pontos (piora) com placebo no Mês 9 (Tabela 1). A mudança favoreceu significativamente Vutrisirana Sódica (Figura 1).
A importância clínica foi avaliada pela mudança no questionário Norfolk Quality of Life - Neuropatia Diabética (QoL-DN). Este questionário (respondido pelo paciente) avalia qualidade de vida relacionada à neuropatia, variando de -4 a 136, onde maior pontuação indica pior qualidade de vida.
O tratamento melhorou em 16,2 pontos no Norfolk QoL-DN versus placebo (p<0,0001), com mudança média de -3,3 pontos (melhora) com Vutrisirana Sódica versus aumento de 12,9 pontos (piora) com placebo no Mês 9 (Tabela 1). A mudança favoreceu significativamente Vutrisirana Sódica (Figura 2).
Outro resultado foi a mudança na velocidade da marcha (teste de caminhada de 10 metros). O tratamento melhorou significativamente versus placebo (Tabela 1).
Avaliações no Mês 18
As avaliações incluíram mudanças até o Mês 18 para mNIS+7, NorfolkQoL-DN, teste de caminhada de 10 metros, estado nutricional (índice de massa corporal modificado [mIMC]) e capacidade de realizar atividades diárias (Rasch-Built Overall Disability Scale [R-ODS]).
Vutrisirana Sódica demonstrou melhorias significativas em todos os resultados (Tabela 1) versus placebo externo (todos p < 0,0001).
A redução mediana nos níveis de TTR no grupo vutrisirana foi não-inferior ao grupo patisirana até o Mês 18 com diferença de 5,3% (IC 95% 1,2; 9,3).
Tabela 1 - Resultados de Eficácia Clínica do Estudo HELIOS-A
| Resultadoa | Início, Média (DP) | Mudança do Início, Média LS (SEM) | Diferença Vutrisirana Sódica- Placebob , Média LS (IC 95%) | Valor p | ||
| Vutrisirana Sódica N=122 | Placebob N=77 | Vutrisirana Sódica | Placebob | |||
| Mês 9 | ||||||
| mNIS+7c | 60.6 (36.0) | 74.6 (37.0) | -2.2 (1.4) | 14.8 (2.0) | -17.0 (-21.8, -12.2) | p<0.0001 |
| Norfolk QoL-DNc | 47.1 (26.3) | 55.5 (24.3) | -3.3 (1.7) | 12.9 (2.2) | -16.2 (-21.7, -10.8) | p<0.0001 |
| Teste de caminhada de 10 metros (m/seg)d | 1.01 (0.39) | 0.79 (0.32) | 0 (0.02) | -0.13 (0.03) | 0.13 (0.07, 0.19) | p<0.0001 |
| Mês 18 | ||||||
| mNIS+7c | 60.6 (36.0) | 74.6 (37.0) | -0.5 (1.6) | 28.1 (2.3) | -28.5 (-34.0, -23.1) | p<0.0001 |
| Norfolk QoL-DNc | 47.1 (26.3) | 55.5 (24.3) | -1.2 (1.8) | 19.8 (2.6) | -21.0 (-27.1, -14.9) | p<0.0001 |
| Teste de caminhada de 10 metros (m/seg)d | 1.01 (0.39) | 0.79 (0.32) | -0.02 (0.03) | -0.26 (0.04) | 0.24 (0.15, 0.33) | p<0.0001 |
| mIMCe | 1057.5 (233.8) | 989.9 (214.2) | 25.0 (9.5) | -115.7 (13.4) | 140.7 (108.4, 172.9) | p<0.0001 |
| R-ODSf | 34.1 (11.0) | 29.8 (10.8) | -1.5 (0.6) | -9.9 (0.8) | 8.4 (6.5, 10.4) | p<0.0001 |
Abreviaturas: IC=intervalo de confiança; LS média dos mínimos quadrados; mIMC=índice de massa corporal modificada; mNIS=Escore modificado de Comprometimento da Neuropatia; QoL-DN = Qualidade de Vida - Neuropatia Diabética; DP = desvio padrão; SEM = erro padrão da média.
a Todos os resultados do Mês 9 analisados com análise de covariância (ANCOVA) com método de imputação múltipla (MI) e todos do Mês 18 com modelo de efeitos mistos para medidas repetidas (MMRM).
b Grupo placebo externo do estudo controlado randomizado APOLLO.
c Número menor indica menos comprometimento/menos sintomas.
d Número maior indica menos incapacidade.
e mIMC: IMC (kg/m2 ) multiplicado por albumina sérica (g/L); número maior indica melhor estado nutricional.
f Número maior indica menor incapacidade.
Figura 1 Alteração no mNIS+7 desde o Início (Mês 9 e Mês 18)
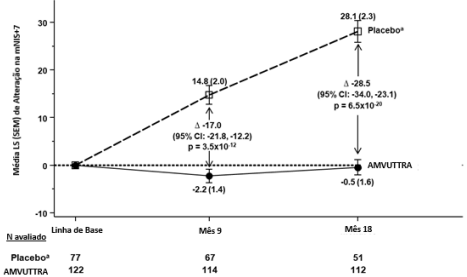
Diminuição no mNIS+7 indica melhora.
Δ indica diferença entre grupos, mostrada como diferença média de LS (IC 95%) para Vutrisirana Sódica – placebo Todos os resultados do Mês 9 analisados com ANCOVA com MI e Mês 18 com MMRM.
a Grupo placebo externo do estudo APOLLO.
Figura 2 Alteração na Pontuação total Norfolk QoL-DN desde o início (Mês 9 e Mês 18)
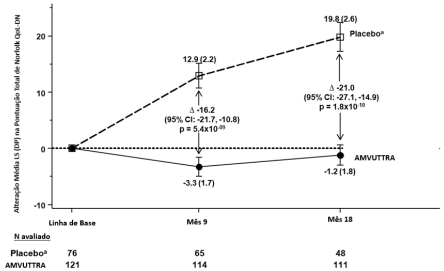
Diminuição na pontuação indica melhora.
Δ indica diferença entre grupos, mostrada como diferença média de LS (IC 95%) para Vutrisirana Sódica – placebo Todos os resultados do Mês 9 analisados com ANCOVA com MI e Mês 18 com MMRM.
a Grupo placebo externo do estudo APOLLO.
Pacientes que receberam Vutrisirana Sódica tiveram melhorias semelhantes versus placebo no mNIS+7 e Norfolk QoL-DN em todos os subgrupos, incluindo idade, sexo, raça, região, escore NIS, status do genótipo V30M, uso anterior de estabilizador de TTR, estágio da doença e pacientes com envolvimento cardíaco pré-definido (Figura 3, 4). As diferenças médias de tratamento foram consistentes entre Mês 9 e Mês 18. O benefício ocorreu independentemente do genótipo (V30M ou não-V30M) e em todos os graus de gravidade.
Figura 3 Forest Plots da Média de Diferença de Tratamento (Mudança do Início até Mês 18), por Subgrupo - mNIS+7
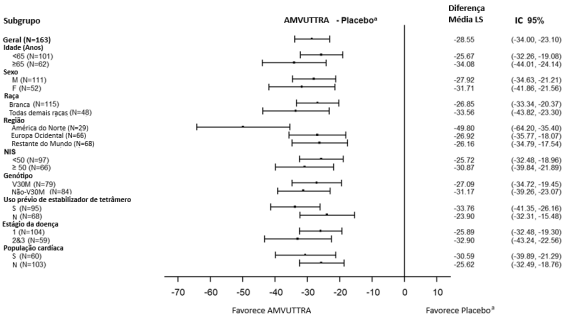
a Grupo placebo externo do estudo APOLLO.
Figura 4 Forest Plots da Média de Diferença de Tratamento (Mudança do Início até Mês 18), por Subgrupo - Pontuação Total Norfolk QoL-DN
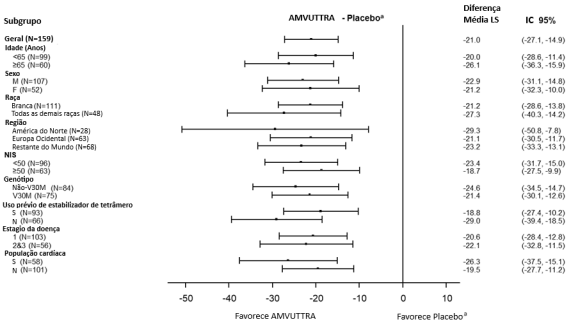
a Grupo placebo externo do estudo APOLLO.
Melhorias versus placebo foram observadas no fragmento N-terminal do pró-hormônio do peptídeo natriurético tipo B (NT-proBNP), biomarcador de disfunção cardíaca. No Mês 18, os níveis médios de NT-proBNP caíram 6% com Vutrisirana Sódica, enquanto aumentaram 96% com placebo.
Ecocardiogramas mostraram alterações na espessura da parede do VE (diferença média LS: -0,18 mm [IC 95% -0,74, 0,38]) e tensão longitudinal (diferença média LS: -0,4% [IC 95% -1,2, 0,4]) com Vutrisirana Sódica versus placebo. Diminuição da espessura e tensão indicam melhorias.
Dada a rápida progressão em pacientes não tratados, os dados apoiam início precoce do tratamento com Vutrisirana Sódica para prevenir progressão da doença.
Características Farmacológicas
Propriedades farmacodinâmicas
Mecanismo de ação
Em pacientes com amiloidose hATTR, as proteínas TTR mutantes e normais formam depósitos amiloides nos tecidos, causando polineuropatia e miocardiopatia progressivas.
Vutrisirana é um siRNA de cadeia dupla modificado que ataca especificamente o RNA mensageiro (mRNA) da TTR.
Através do RNA de interferência (RNAi), a vutrisirana degrada o mRNA da TTR no fígado, reduzindo a proteína TTR no sangue e diminuindo depósitos amiloides.
Características farmacodinâmicas
No estudo de Fase 3 HELIOS-A, os efeitos de 25 mg de Vutrisirana Sódica aplicados sob a pele a cada 3 meses foram avaliados. A TTR sérica média reduziu 64% no Dia 22, com redução de 73% na Semana 6. Com aplicações repetidas, reduções médias após 9 e 18 meses foram 83% e 88%.
Reduções semelhantes ocorreram independente de genótipo, uso anterior de estabilizador de TTR, peso, sexo, idade ou raça.
Vutrisirana Sódica diminuiu os níveis de vitamina A, com reduções médias de pico e vale de 70% e 63%.
Eletrofisiologia Cardíaca
Vutrisirana não afetou o intervalo QTc em pessoas saudáveis com doses até 300 mg. Não foi feito estudo QT completo.
Propriedades farmacocinéticas
As propriedades farmacocinéticas foram caracterizadas medindo concentrações plasmáticas e urinárias de vutrisirana.
Absorção
Após aplicação sob a pele, vutrisirana é absorvida rapidamente com tempo até concentração máxima (tmax) de 3,0 horas. Na dose de 25 mg a cada 3 meses, as concentrações máximas médias e área sob a curva foram 0,12 μg/mL e 0,80 μg·h/mL. Não houve acúmulo no plasma após doses repetidas.
Distribuição
Vutrisirana liga-se a proteínas plasmáticas em mais de 80%. A ligação diminuiu com concentrações maiores. O volume estimado de distribuição foi 10,2 L. Vutrisirana distribui-se principalmente para o fígado após aplicação.
Metabolismo
Vutrisirana é metabolizada por enzimas em fragmentos no fígado. Não havia metabólitos circulantes importantes. Estudos in vitro indicam que não sofre metabolização por enzimas CYP450.
Eliminação
Após dose única de 25 mg, a depuração plasmática mediana foi 21,4 L/h. A meia-vida terminal mediana foi 5,23 horas. Após doses únicas, 15,4 a 25,4% foi eliminado inalterado na urina.
Linearidade/não linearidade
Após doses únicas de 5 a 300 mg, a concentração máxima foi proporcional à dose.
Relação farmacocinética/farmacodinâmica
Análises mostraram relação dose-dependente entre concentrações hepáticas previstas e reduções na TTR sérica. Reduções médias previstas foram 88% (pico), 86% (vale) e 87% (média). A análise indicou redução similar em pacientes com insuficiência renal leve a moderada ou hepática leve, sexo, raça, uso anterior de estabilizadores, genótipo, idade e peso.
Populações especiais
Sexo e Raça
Estudos não mostraram diferenças clinicamente significativas na farmacocinética ou redução de TTR por sexo ou raça.
Problemas no Fígado
Análises não indicaram impacto em casos leves na exposição ou redução de TTR versus função normal. Não foi estudado em casos moderados ou graves.
Problemas nos Rins
Análises não indicaram impacto em casos leves ou moderados na exposição ou redução de TTR versus função normal. Não foi estudado em casos graves.
Pacientes Pediátricos
Segurança e eficácia não foram estudadas em menores de 18 anos.
Pacientes idosos
No estudo HELIOS-A, 38% dos pacientes tratados tinham ≥65 anos (5,7% ≥75). Não houve diferenças significativas na farmacocinética ou redução de TTR entre pacientes <65 e ≥65 anos.
Dados de segurança pré-clínicos
Farmacologia Animal/Toxicologia
Vutrisirana é farmacologicamente ativa em macacos, mas não em roedores ou coelhos.
Em macacos, doses únicas sob a pele de 0,3 e 1 mg/kg reduziram TTR sérica em 60% e 95%. Doses repetidas mensais a 1 e 3 mg/kg reduziram TTR >95%. Doses ≥30 mg/kg reduziram TTR (até 99%) e vitamina A (até 89%) sem achados tóxicos. Exames oculares foram normais.
Não houve efeitos nos sistemas cardiovascular, respiratório ou nervoso central em macacos.
Em estudos de toxicidade com doses repetidas mensais em ratos e macacos, nenhum órgão-alvo foi identificado nas maiores doses testadas.
Com base na ausência de resultados, vutrisirana não tem potencial imunoestimulador ou de imunotoxicidade.
Carcinogenicidade
Em estudo de 2 anos em ratos, vutrisirana não foi cancerígena nas maiores doses testadas.
Genotoxicidade
Vutrisirana não foi mutagênica em ensaios.
Teratogenicidade
Em estudo com ratas grávidas, doses ≤30 mg/kg não causaram malformações.
Em estudo com coelhas grávidas, doses ≤30 mg/kg não causaram efeitos adversos.
Em estudo de desenvolvimento, doses ≤20 mg/kg não afetaram crescimento da prole.
Fertilidade
Doses semanais até 70 mg/kg em ratos não afetaram fertilidade.
Armazenamento do Amvuttra
Guarde em temperatura ambiente (até 30°C) ou geladeira (2°C a 8°C). Não congele.
Mantenha na embalagem original até o uso.
Lote e datas de fabricação/validade: veja na embalagem.
Não use após o vencimento. Guarde na embalagem original.
Características do medicamento
Este remédio é uma solução clara, de incolor a amarela. Cada caixa contém uma seringa pronta de uso único.
O componente principal é vutrisirana sódica. Cada seringa contém 25 mg de vutrisirana em 0,5 mL.
Outros componentes: fosfato de sódio monobásico di-hidratado, fosfato de sódio dibásico di-hidratado, cloreto de sódio, água para injetáveis, hidróxido de sódio e ácido fosfórico.
Antes de usar, verifique o aspecto do remédio. Se houver mudança, consulte o farmacêutico.
Mantenha fora do alcance de crianças.
Dizeres Legais do Amvuttra
MS - 1.9361.0004.001-6
Farmacêutico Responsável:
Marcelo Chaves de Oliveira
CRF-GO 5339
Registrado e Importado por:
Specialty Pharma Goias Ltda.
Av Segunda Avenida Quadra 01B lote 48-E 6º andar sala 616-620
Aparecida de Goiania, GO, Brasil
CNPJ 31.731.807/0001-28
Fabricado por:
Vetter Pharma-Fertigung GmbH & Co. KG
Ravensburg, Alemanha
Embalado por:
Sharp Packaging Services, LLC.
Allentown, Estados Unidos
SAC
0800-0474597
Venda sob prescrição médica.